Article Text

Abstract
Objectives To investigate the clinical impact of 1-year certolizumab pegol (CZP) therapy added to the first year of 2-year methotrexate (MTX) therapy, compared with 2-year therapy with MTX alone.
Methods MTX-naïve patients with early rheumatoid arthritis (RA) with poor prognostic factors were eligible to enter Certolizumab-Optimal Prevention of joint damage for Early RA (C-OPERA), a multicentre, randomised, controlled study, which consisted of a 52-week double-blind (DB) period and subsequent 52-week post treatment (PT) period. Patients were randomised to optimised MTX+CZP (n=159) or optimised MTX+placebo (PBO; n=157). Following the DB period, patients entered the PT period, receiving MTX alone (CZP+MTX→MTX; n=108, PBO+MTX→MTX; n=71). Patients who flared could receive rescue treatment with open-label CZP.
Results 34 CZP+MTX→MTX patients and 14 PBO+MTX→MTX patients discontinued during the PT period. From week 52 through week 104, significant inhibition of total modified total Sharp score progression was observed for CZP+MTX versus PBO+MTX (week 104: 84.2% vs 67.5% (p<0.001)). Remission rates decreased after CZP discontinuation; however, higher rates were maintained through week 104 in CZP+MTX→MTX versus PBO+MTX→MTX (41.5% vs 29.3% (p=0.026), 34.6% vs 24.2% (p=0.049) and 41.5% vs 33.1% (p=0.132) at week 104 in SDAI, Boolean and DAS28(erythrocyte sedimentation rate) remission. CZP retreated patients due to flare (n=28) showed rapid clinical improvement. The incidence of overall adverse events was similar between groups.
Conclusions In MTX-naïve patients with early RA with poor prognostic factors, an initial 1 year of add-on CZP to 2-year optimised MTX therapy brings radiographic and clinical benefit through 2 years, even after stopping CZP.
Trial registration number NCT01451203.
- Early Rheumatoid Arthritis
- DMARDs (biologic)
- Methotrexate
- Anti-TNF
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease characterised by progressive inflammatory synovitis. This results in the destruction of articular cartilage and marginal bone, which is generally thought to be irreversible.1 Recent studies have demonstrated that the early treatment of patients with antirheumatic drugs is associated with a reduction in inflammation, greater inhibition of structural damage and better long-term outcomes.2 ,3 Furthermore, early aggressive treatment with biological disease-modifying antirheumatic drugs (bDMARDs), such as antitumour necrosis factors (TNFs), was reported to be highly effective at reducing disease progression.4 The effect of treatment discontinuation/tapering following successful inhibition of disease progression as a result of using bDMARDs early in the course of the disease has yet to be fully investigated; however, there is the possibility that the positive disease trajectory may be maintained following treatment cessation.
Certolizumab pegol (CZP) is a humanised anti-TNF antibody fragment conjugated to polyethylene glycol, approved for the treatment of inflammatory diseases, including RA. The efficacy and safety of CZP in combination with methotrexate (MTX) during the early stages of RA was assessed in the Certolizumab-Optimal Prevention of joint damage for Early RA (C-OPERA) study. This study consisted of two periods: a 52-week double-blind (DB) period during which patients received either CZP or placebo (PBO) together with MTX, and a subsequent 52-week post-PBO/CZP treatment (PT) period in which patients received MTX therapy without CZP or PBO. Results from the DB period, which showed significant inhibition of structural damage and a reduction in the severity of RA symptoms following treatment with CZP+MTX compared with PBO+MTX, have been reported.4 Here, we report the 2-year overall results including the PT period, which investigated whether the clinical benefits of initial 1-year CZP+MTX therapy were sustained through a subsequent 1-year period where patients received MTX alone.
Methods
Patients
The eligibility criteria for the DB period were previously reported.4 Brief details are provided in the online supplementary materials.
supplementary materials
Study design
C-OPERA (NCT01451203) was a multicentre, DB, PBO-controlled, randomised, parallel-group study conducted in Japan and was previously described.4 Full details of the study design are provided in the online supplementary materials.
Efficacy assessments
Efficacy assessments were performed every 8 weeks during the PT period. The primary analysis of the PT period was change in van der Heijde modified total Sharp score (mTSS) from baseline at week 104, which compared the progression of joint damage between the CZP+MTX→MTX and PBO+MTX→MTX groups. Inhibition of joint damage progression was assessed using mTSS at weeks 0, 52 and 104; mTSS was evaluated by two readers independently, in accordance with previously reported methods.5 ,6 Additional analyses comparing clinical efficacy between the two groups included disease activity score (DAS)28(erythrocyte sedimentation rate (ESR)), simple disease activity index (SDAI), swollen joint count (SJC), tender joint count (TJC), Health Assessment Questionnaire Disability Index (HAQ-DI), physician's and patient's global assessments of disease activity (PtGADA), patient's assessment of arthritis pain, ESR and C-reactive protein (CRP) levels. Clinical remission was defined as achieving SDAI ≤3.3, DAS28(ESR) <2.6 or ≤1 on all four of the following criteria (Boolean remission): the number of TJC (in 28 joints), number of SJC (in 28 joints), CRP (mg/dL) and PtGADA (100 mm visual analogue scale (VAS) data converted to cm).
Safety assessments
All safety events during the PT period were recorded as adverse events (AEs) or serious AEs (SAEs). Laboratory tests (haematological, blood chemistry, urinalysis), chest radiographs and ECG were also evaluated.
Statistical analyses
Full details of the statistical analyses can be found in the online supplementary materials. In brief, the full analysis set (FAS; defined as all patients who received ≥1 dose of study drug and provided any efficacy data thereafter) was used for all efficacy measurements. Missing data were imputed using linear extrapolation for mTSS and last observation carried forward (LOCF) for all other efficacy variables. Change from baseline in mTSS at weeks 52 and 104 was analysed using an analysis of covariance (ANCOVA) model. Fisher's exact test was used to compare rates of mTSS non-progression (mTSS change from baseline ≤0.5) and clinical remission at weeks 52 and 104, between the PBO and CZP groups.
Results
Patient characteristics and disposition
Of the 316 patients who were randomised and received at least one dose of study drug (FAS population), 179 patients entered the PT period and 131 patients completed the study (figure 1). The proportion of patients completing the PT period (from the patients who entered the PT period) was 68.5% and 80.3% in the CZP+MTX→MTX group and the PBO+MTX→MTX group, respectively (figure 1).

Patient disposition in the Certolizumab-Optimal Prevention of joint damage for Early rheumatoid arthritis trial. CZP, certolizumab pegol; FAS, full analysis set; MTX, methotrexate; PBO, placebo.
Patient baseline demographics and disease characteristics at DB baseline and at PT period entry (week 52) are shown in table 1 and online supplementary table S1. At DB baseline, patient demographics and disease characteristics were similar between the groups. In both groups, disease activity at baseline in the PT population was slightly lower than in the total population, and decreased from baseline through week 52 following treatment with either CZP+MTX or PBO+MTX.
Baseline demographics and patient characteristics
Inhibition of joint damage progression in the total population
As reported previously, at week 52 the change from baseline in mTSS was smaller in the CZP+MTX→MTX group compared with the PBO+MTX→MTX group (0.36±2.70 vs 1.58±4.86, p<0.001 by ANCOVA), and the rate of radiographic non-progression was higher in the CZP+MTX→MTX group compared with the PBO+MTX→MTX group (82.9% vs 70.7%; p<0.011 by Fisher's exact test).4 During the PT period, through week 104, the changes from baseline (mean±SD) for total mTSS (0.66±5.38 vs 3.01±9.66 (p=0.001)), erosion score (0.30±2.54 vs 1.43±4.40 (p=0.003)) and joint space narrowing score (0.36±4.27 vs 1.58±7.17 (p=0.002)) were lower for the CZP+MTX→MTX group compared with the PBO+MTX→MTX group using linear extrapolation for missing data imputation (figure 2A). A sensitivity analysis using an LOCF imputation method (figure 2A) confirmed the results of the primary analysis (linear extrapolation). At week 104, the proportion of patients with radiographic non-progression (ie, mTSS change from baseline ≤0.5) was higher for the CZP+MTX→MTX group compared with the PBO+MTX→MTX group (84.2% vs 67.5%, p<0.001). Furthermore, the proportion of patients with rapid radiographic progression (RRP: mTSS yearly change from baseline ≥5) at week 104 was lower for CZP+MTX→MTX group compared with the PBO+MTX→MTX group (3.2% vs 9.6%, p=0.022). Subgroup analyses revealed that high baseline mTSS, CRP or TNF was associated with poor week 104 radiographic outcomes in the PBO+MTX→MTX group. The CZP+MTX→MTX group also showed higher inhibition of radiographic progression in these populations (see online supplementary table S3). Consistent with radiographic findings, the proportion of patients with HAQ remission (HAQ ≤0.5) at week 104 was numerically higher in the CZP+MTX→MTX group than the PBO+MTX→MTX group (73.0% vs 63.7%, p=0.09). In addition, the proportion of the patients who achieved HAQ remission at week 104 was higher in patients who showed non-radiographic progression at week 104 than those who did not (76.7% vs 52.0% (p=0.015) in CZP+MTX→MTX, and 70.8% vs 49.0% (p=0.013) in PBO+MTX→MTX).

(A) Change from baseline in modified total Sharp score (mTSS) at week 104. (B) Cumulative probability plot of mTSS change from baseline at week 104 for both PBO+MTX→MTX and CZP+MTX→MTX groups. Change from baseline in mTSS was analysed using an analysis of covariance model in which actual scores were converted to rank scores, using the treatment group as a factor and baseline rank score as a covariate. Rate of mTSS non-progression (mTSS change from baseline ≤0.5) was compared using Fisher's exact test. CZP, certolizumab pegol; LOCF, last observation carried forward; MTX, methotrexate; PBO, placebo.
Clinical remission in the total population
The numbers of patients achieving SDAI, Boolean and DAS28(ESR) remission were calculated throughout both the DB and PT periods (figure 3). At the end of the DB period (week 52), clinical remission rates were significantly higher in the CZP+MTX→MTX group compared with the PBO+MTX→MTX group.4

The proportion of patients achieving (A) SDAI remission, (B) Boolean remission and (C) DAS28(ESR) remission during the Certolizumab-Optimal Prevention of joint damage for Early rheumatoid arthritis study in both the PBO+MTX→MTX and CZP+MTX→MTX groups. Remission rates at weeks 52 and 104 were compared using Fisher's exact test. CZP, certolizumab pegol; DB, double blind; ESR, erythrocyte sedimentation rate; MTX, methotrexate; PBO, placebo; PT, post treatment.
The rates of clinical remission observed for the CZP+MTX→MTX group decreased during the first 16 weeks of the PT period; however, the rate stabilised from week 68 (week 16 of the PT period) through week 104. The remission rates of the PBO+MTX→MTX group during the PT period were similar to the DB period, with no change in clinical remission observed during the transition from the end of DB to the PT period. Throughout the duration of the PT period, the rates of clinical remission remained higher in the CZP+MTX→MTX group compared with the PBO+MTX→MTX group (41.5% vs 29.3% (p=0.026), 34.6% vs 24.2% (p=0.049) and 41.5% vs 33.1% (p=0.132) at week 104 in SDAI, Boolean and DAS28(ESR), respectively). The proportion of patients with low disease activity (DAS28(ESR) ≤3.2) was also higher in the CZP+MTX→MTX group compared with the PBO+MTX→MTX group throughout the PT period (see online supplementary figure S1).
Impact of CZP discontinuation in the CZP+MTX→MTX group
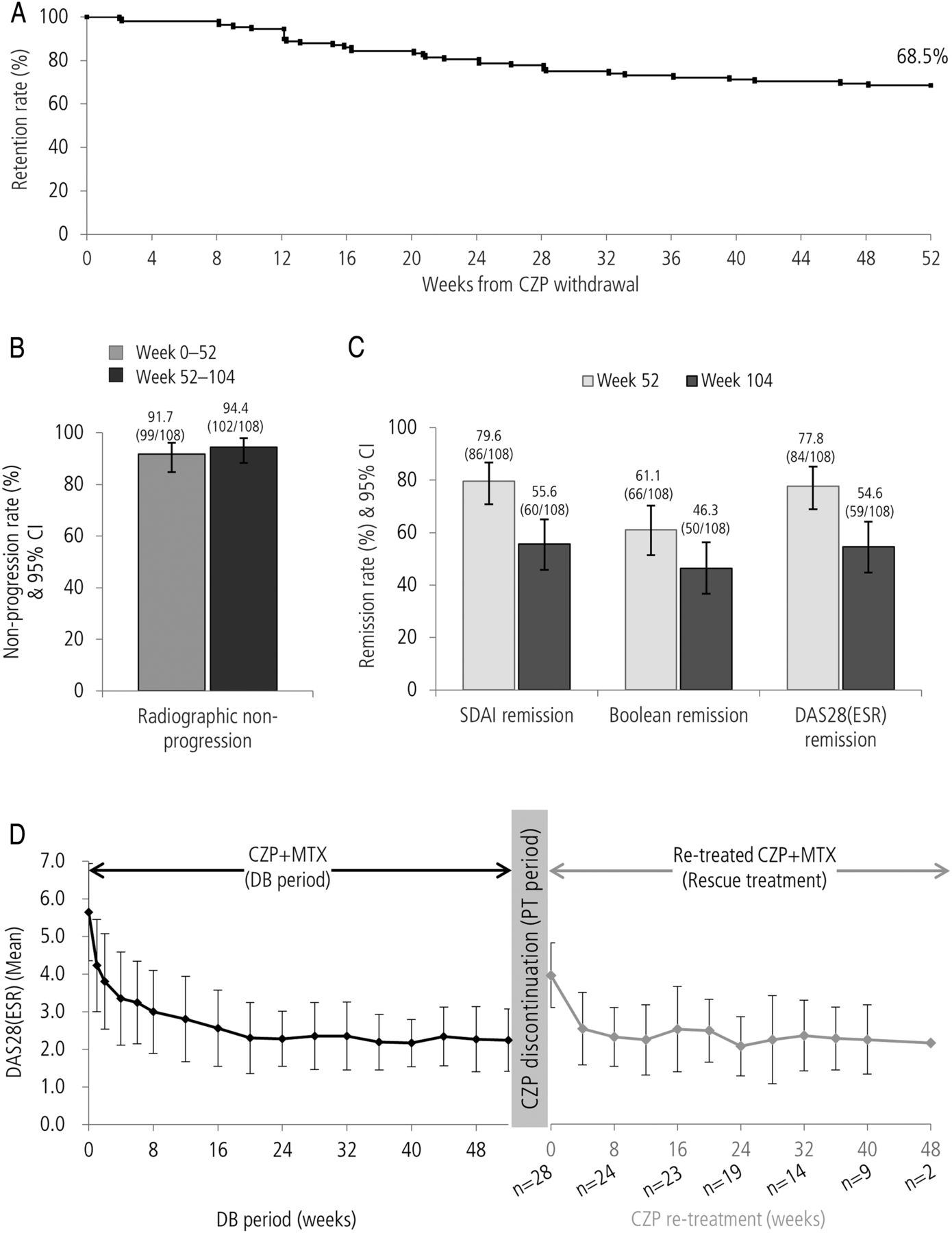
The impact of CZP discontinuation was assessed on patients who entered the PT period (PT population) from the CZP+MTX group (n=108). Of these, 74 patients (68.5%) completed the 1-year PT period with MTX therapy (figure 4A). Rates of radiographic non-progression during the PT period (94.4% (102/108); figure 4B) were similar to the rates observed during the DB period (91.7% (99/108)), as were mean changes in mTSS (±SD) during the first and second 52-week periods (DB period (weeks 0−52): −0.03 (±0.80); PT period (weeks 52−104): 0.06 (±0.76)). Conversely, clinical remission rates in the PT population of the CZP+MTX→MTX group showed decreases in SDAI, Boolean and DAS28(ESR) definitions of remission from weeks 52 to 104 (from 79.6% to 55.6%, 61.1% to 46.3%, 77.8% to 54.6%, respectively; figure 4C).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Retention rate after discontinuation of CZP (Kaplan-Meier plot), (B) clinical remission at weeks 52 and 104, (C) DAS28(ESR) over time during DB period and after CZP restart in patients who were retreated with CZP (n=28), (D) modified total Sharp score non-progression rate during and after 52-week CZP therapy for patients who entered the PT period from CZP+MTX group. CZP, certolizumab pegol; DB, double blind; ESR, erythrocyte sedimentation rate; MTX, methotrexate; PT, post treatment.
Retreatment of CZP after symptom flare in CZP+MTX→MTX group
Of the 34 patients who withdrew during the PT period, 28 patients moved to rescue treatment, restarting CZP therapy as a consequence of symptom flares (figure 1). Improvements were observed in this cohort of patients (figure 4D); mean disease activity decreased from DAS28(ESR) 4.40 at CZP restart (n=28) to 2.49 after 12 weeks (n=25; observed case). Of the 28 patients receiving rescue treatment, 26 continued retreatment until the end of the study. The majority of patients receiving CZP retreatment for ≥12 weeks (n=25) responded positively; using DAS28(ESR) criteria, 24/25 achieved low disease activity and 21/25 achieved remission on at least one study visit.
Safety
Study drug exposure during both the total study period and the PT period was higher for the CZP+MTX→MTX group (total: 223.6 patient-years (PY), PT: 87.7 PY) compared with the PBO+MTX→MTX group (total: 179.4 PY, PT: 63.4 PY; table 2). This difference could be attributed to the higher withdrawal rate in the PBO+MTX→MTX group. Overall, no clinically relevant difference was observed in the total incidence of AEs between the CZP+MTX→MTX group and the PBO+MTX→MTX group through week 104 (154 patients (96.9%) vs 150 patients (95.5%)), or SAEs (17 patients (10.7%) vs 18 patients (11.5%)). Incidence rates of some types of AEs including infections and infestations, pneumonia and hepatic disorders were higher during the DB period (weeks 0–52) compared with the PT period (weeks 52–104); however, this increase was observed for both the CZP+MTX→MTX and the PBO+MTX→MTX group (table 2 and see online supplementary table S2).
Summary of treatment-emergent adverse events (TEAE)
Discussion
Biological DMARDs are considered second-line therapies for patients who cannot achieve treatment targets using conventional synthetic disease-modifying antirheumatic drugs (csDMARDs) in the management of RA.7 However, it has been reported that the inhibitory effect of bDMARDs on joint damage is superior to that of csDMARDs, including MTX.4 ,8 Although there is some evidence of bone erosion repair following treatment with bDMARDs,9 joint destruction in patients with RA is generally considered to be irreversible.10 Consequently, prevention of significant joint damage is crucial to avoid permanent functional disability,11 supporting early treatment with bDMARDs.
Concerns have been raised that initiating aggressive treatment with a bDMARD may be excessive for some patients and so identifying patients who would particularly benefit from initial aggressive treatment is critical when considering it. The feasibility of bDMARD withdrawal after achieving a therapeutic target is also of importance from both safety and economical points of view. If these issues are overcome, there is the possibility of a clinical approach where RA therapy is initiated with a bDMARD in the early stage of disease, leading to improved outcomes that can be maintained even after withdrawal of the initial aggressive treatment.12
C-OPERA was designed to assess the clinical benefit of CZP treatment concomitant with MTX as first-line therapy for early RA, particularly for patients who were considered to require aggressive treatment. Patients who had poor prognostic factors, including a high titre of anticyclic citrullinated peptide antibody in addition to either rheumatoid factor positivity or bone erosions, were eligible to enter the study. C-OPERA was a study composed of two periods. The results from the first year of the study demonstrated the clinical benefit of adding CZP to MTX therapy (DAS28(ESR) remission and radiographic non-progression was achieved in more than 50% and 80% of patients, respectively), suggesting that the introduction of CZP at a very early stage led to substantial therapeutic effects, despite poor prognosis.4 In this report, we assessed whether the clinical benefit of initial 1-year CZP+MTX treatment was observed after stopping CZP and continuing with MTX therapy for 1 year.
A key finding from the PT period reported here, was that the beneficial effect seen after an initial 1 year of treatment with CZP was observed after discontinuing CZP therapy when the dose of MTX remained optimised. Radiographic progression, estimated using linear extrapolation, remained lower in the CZP+MTX→MTX group compared with the PBO+MTX→MTX group. When using linear extrapolation, patient withdrawal can result in an overestimation of mTSS change from baseline. To overcome this, analyses were repeated using LOCF imputation, confirming the efficacy of CZP co-administration. The rate of radiographic non-progression observed in the CZP+MTX→MTX patient population during the PT period was similar to that observed during the DB period in the same population. This suggests that joint destruction may be prevented even after discontinuation of CZP in patients who responded adequately to initial treatment with CZP+MTX. These data, combined with the lower rate of RRP in the CZP+MTX→MTX group compared with the PBO+MTX→MTX, and the subgroup analyses (see online supplementary table S3) suggest that induction treatment with CZP+MTX benefits those with a high risk of radiographic progression. The rate of HAQ remission was higher in patients with radiographic non-progression compared with those with radiographic progression, suggesting that radiographic progression associates with functional disability even during the 2 years of the study. These results suggest that early treatment with CZP during the initial stages of the disease, when rapid joint damage may take place,13 could prevent long-term progression of joint damage and functional disability as previously suggested in the ‘window of opportunity’ concept.8 ,14–16
In addition to joint damage prevention, the rates of clinical remission throughout the PT period remained higher in the CZP+MTX→MTX group compared with the PBO+MTX→MTX group. After CZP discontinuation, approximately 25% of the patients flared; however, they showed rapid response to CZP retreatment, with recovery to preflare disease activity levels. Although joint destruction was consistently prevented in the PT population following CZP withdrawal, clinical remission was sometimes lost. Discrepancies in clinical and radiographic efficacy have been reported for adalimumab (ADA);17 similar differences in the clinical and radiographic efficacies of CZP that continue following treatment discontinuation could be responsible for the results observed here. A decrease in remission rate was mainly observed during the first 16 weeks after CZP discontinuation. We speculate that patients who still produce excessive TNF may have flared as the concentration of CZP decreased.
Overall, the results suggest that initial aggressive treatment with a bDMARD could be a potential treatment option at the early stage of disease, especially for patients who have a poor prognosis. Once patients achieve their treatment targets, bDMARD treatment could be withdrawn. This treatment approach has the potential to prevent irreversible joint damage, reduce patient risk of AEs and be a more cost-effective way to manage RA over the long term. This approach is supported by two further studies. The Optimal Protocol for Methotrexate and Adalimumab Combination Therapy in Early Rheumatoid Arthritis (OPTIMA) study demonstrated minimal loss of clinical response without significant radiographic progression after ADA withdrawal in patients with early RA who initiated combination therapy with MTX.8 While, The High Induction Therapy with Anti-Rheumatic Drugs (HIT HARD) study showed better radiographic outcomes but no significant difference in disease activity after ADA withdrawal in the ADA+MTX→MTX group compared with MTX alone.18 Differences in the results of these studies compared with C-OPERA suggest that the condition of the ‘induction-maintenance’ regimen12 may be important. Further studies are now needed to identify the appropriate disease activity targets required to achieve continued disease inhibition following CZP withdrawal, and to identify patient populations that would particularly benefit from first-line therapy with CZP. Moreover, additional analyses are also required to confirm whether this approach has significant clinical benefits for patients failing to respond to MTX therapy after 3 months, which is the approach currently recommended in treat-to-target guidelines.19
Safety analyses showed similar rates of SAEs for both the CZP+MTX→MTX and PBO+MTX→MTX groups over the 2 years of the C-OPERA study, indicating that there are no major safety concerns when adding CZP to optimised MTX therapy. Incidences of AEs and SAEs during the PT period were lower compared with the initial DB period in both groups. One reason for this may be ‘survival bias’, where patients discontinued the study because of an intolerance to the study drugs (CZP and/or MTX) in the first period, resulting in a lower AE rate in the second.20
This study has several limitations. In clinical practice, only patients failing to respond to MTX would receive CZP therapy, and so it is not known how this approach compares with initial CZP therapy. No patients received CZP for a full 2 years or were treated with a reduced dose of CZP, so it was not possible to compare these treatment regimens with CZP discontinuation. CZP withdrawal had not been optimised; therefore, there is the potential for further investigation regarding the appropriate treatment targets and the timing of CZP withdrawal in different patient populations. There were differences between the study design of C-OPERA and current RA treatment recommendations. For example, in clinical practice, treatment recommendations for patients with poor prognostic factors include using additional DMARDs in addition to MTX,7 ,21 which was prohibited in C-OPERA. C-OPERA was not designed as an intercontinental global study; thus, it is not known whether these results are generalisable to ethnicities other than Japanese. In particular, the MTX dose of 16 mg is low compared with similar RA studies from the European Union and USA (15−17 mg/week).22–26 However, when considering differences in patient body weight and MTX metabolism,27 a lower dose of MTX in this study may correspond to the doses used in those previous studies. Finally, as non-responding patients were eligible to receive rescue treatment from 24 weeks onwards, we cannot exclude the possibility that a proportion of the 70 PBO+MTX patients switching to rescue therapy in the first year may have achieved clinical response if treated for a longer period of time.4
Overall, these results suggest that patients with early RA would benefit from the addition of CZP to MTX therapy during the early stages of disease, particularly with respect to the prevention of joint destruction. Although this aggressive therapeutic strategy would not be recommended for all patients, it may be a potential option for those patients with a high risk of experiencing rapid joint destruction. How to identify these patients at disease onset requires further investigation.
Acknowledgments
The authors acknowledge Lilia Marinova MD, PhD, UCB Pharma, UK, for publication coordination, and Simon Foulcer, PhD, and Danielle Machin, BSc, from Costello Medical Consulting, Cambridge, UK, for medical writing and editorial assistance in preparing this manuscript for publication, based on the authors' input and direction. UCB Pharma reviewed only for scientific and legal accuracy.
References
Footnotes
Handling editor Tore K Kvien
Contributors All authors were involved in the C-OPERA study, reviewed and interpreted the data, developed the manuscript and approved the final draft.
Funding Astellas Pharma. and UCB Pharma funded this study and manuscript.
Competing interests TA has taken part in speakers' bureaus for Astellas, Bristol-Myers, Chugai and Mitsubishi-Tanabe. KY has received consultancy fees from Abbott, BMS, Chugai, Eisai, Mitsubishi-Tanabe, Pfizer, Roche and UCB Pharma; and has received research grants from Abbott, Eisai, Mitsubishi-Tanabe, Pfizer, Santen and UCB Pharma. TT has received consultancy fees from AstraZeneca, Asahi Kasei, Eli Lilly, Mitsubishi-Tanabe and Novartis; research grants from Abbott, Astellas, BMS, Chugai, Daiichi-Sankyo, Eisai, Janssen, Mitsubishi-Tanabe, Nippon Shinyaku, Otsuka, Pfizer, Sanofi-Aventis, Santen, Takeda and Teijin; and has taken part in speakers' bureaus for Abbott, BMS, Chugai, Eisai, Janssen, Mitsubishi-Tanabe, Pfizer and Takeda and UCB Pharma. HY has received consultancy fees from Abbott, Astellas, BMS, Chugai, Eisai, Janssen, Mitsubishi-Tanabe, Pfizer, Takeda and UCB Pharma; and has received research grants from Abbott, Astellas, BMS, Chugai, Eisai, Janssen, Mitsubishi-Tanabe, Pfizer, Takeda and UCB Pharma. NI has received research grants from Abbott, Astellas, BMS, Takeda, Chugai, Eisai, Janssen, Kaken Mitsubishi-Tanabe and Pfizer; and has taken part in speakers' bureaus for Abbott, Astellas, BMS, Chugai, Eisai, Janssen, Kaken, Mitsubishi-Tanabe, Otsuka, Pfizer, Taisho-Toyama and Takeda. YT has received research grants from Astellas, Abbvie, BMS, Chugai, Daiichi-Sankyo, Mitsubishi-Tanabe, MSD; has received consultancy fees from Abbott, Abbvie, Asahi Kasei, Astellas, AstraZeneca, Chugai, Daiichi-Sankyo, Eisai, Eli Lilly, GSK, Janssen, Mitsubishi-Tanabe, MSD, Pfizer, Quintiles, Takeda and UCB Pharma; and has taken part in speakers' bureaus for Abbott, Abbvie, Asahi Kasei, Astellas, AstraZeneca, Chugai, Daiichi-Sankyo, Eisai, Eli Lilly, GSK, Janssen, Mitsubishi-Tanabe, MSD, Pfizer, Quintiles, Takeda and UCB Pharma. KE has received consultancy fees from UCB Pharma. AW has received research grants from Daiichi-Sankyo, Dainippon-Sumitomo, Kyorin, Meiji Seika; Shionogi, Taiho, Taisho and Toyama Chemical; and has taken part in speakers' bureaus for Daiichi-Sankyo, Dainippon-Sumitomo, GSK, Mitsubishi-Tanabe, MSD, Pfizer, Shionogi and Taisho-Toyama. HO has received consultancy fees from Astellas and UCB Pharma. SY has received research grant from BMS and taken part in speakers' bureaus for Abbvie, Astellas, Chugai, Eizai, Pfizer, Mitsubishi-Tanabe and Takeda. YY has no competing interests to disclose. YK has received speakers' bureau from Astellas, Chugai and Ono. TM has received speaker honoraria from Pfizer Japan, Janssen Pharmaceutical Co.. and Astellas Pharma; and research grants form Quintiles Transnational Japan K.K, Janssen Pharmaceutical Co., Takeda Chemical Industries, Daiichi Sankyo Co., Astellas Pharma, Eli Lilly Japan K.K., MSD Co., Nippon Kayaku Co., Parexel International, Pfizer Japan and Bristol-Myers Squibb. MI has received payment for lectures from Astellas, Chugai, Ono and Tanabe-Mitsubishi; has received research grants from Pfizer and a royalty fee from Chugai. TS is an employee of UCB Pharma; TO is an employee of Astellas. DvdH has received consultancy fees from Abbvie, Amgen, AstraZeneca, Augurex, BMS, Boehringer Ingelheim, Celgene, Centocor, Chugai, Covagen, Daiichi-Sankyo, Eli-Lilly, Galapagos, GSK, Janssen Biologics, Merck, Novartis, Novo-Nordisk, Otsuka, Pfizer, Roche, Sanofi-Aventis, Schering-Plough, UCB Pharma and Vertex; and is the Director of Imaging Rheumatology bv. NM has received research grants from Abbott, Astellas, Chugai, Eisai, Mitsubishi-Tanabe, Pfizer and Takeda. TK has received consultancy fees from Abbvie, Astellas, BMS, Chugai, Daiichi-Sankyo, Eisai, Mitsubishi-Tanabe, Pfizer, Santen, Taisho-Toyama, Takeda, Teijin and UCB Pharma, and has taken part in speakers' bureaus for Abbott, Astellas, BMS, Chugai, Daiichi-Sankyo, Eisai, Mitsubishi-Tanabe, Pfizer, Santen, Taisho-Toyama, Takeda, Teijin and UCB Pharma.
Ethics approval This study was conducted after review and approval by the institutional review board designated by each study site after consideration of the ethical, scientific and medical justification for the conduct of the study.
Provenance and peer review Not commissioned; externally peer reviewed.