Article Text

Abstract
Objectives To characterise the clinical features, immune manifestations and molecular mechanisms in a recently described autoinflammatory disease caused by mutations in TRNT1, a tRNA processing enzyme, and to explore the use of cytokine inhibitors in suppressing the inflammatory phenotype.
Methods We studied nine patients with biallelic mutations in TRNT1 and the syndrome of congenital sideroblastic anaemia with immunodeficiency, fevers and developmental delay (SIFD). Genetic studies included whole exome sequencing (WES) and candidate gene screening. Patients’ primary cells were used for deep RNA and tRNA sequencing, cytokine profiling, immunophenotyping, immunoblotting and electron microscopy (EM).
Results We identified eight mutations in these nine patients, three of which have not been previously associated with SIFD. Three patients died in early childhood. Inflammatory cytokines, mainly interleukin (IL)-6, interferon gamma (IFN-γ) and IFN-induced cytokines were elevated in the serum, whereas tumour necrosis factor (TNF) and IL-1β were present in tissue biopsies of patients with active inflammatory disease. Deep tRNA sequencing of patients’ fibroblasts showed significant deficiency of mature cytosolic tRNAs. EM of bone marrow and skin biopsy samples revealed striking abnormalities across all cell types and a mix of necrotic and normal-appearing cells. By immunoprecipitation, we found evidence for dysregulation in protein clearance pathways. In 4/4 patients, treatment with a TNF inhibitor suppressed inflammation, reduced the need for blood transfusions and improved growth.
Conclusions Mutations of TRNT1 lead to a severe and often fatal syndrome, linking protein homeostasis and autoinflammation. Molecular diagnosis in early life will be crucial for initiating anti-TNF therapy, which might prevent some of the severe disease consequences.
- autoinflammation
- congenital sideroblastic anemia with immunodeficiency
- fevers
- developmental delay (SIFD)
- tRNA
- TRNT1
- TNF inhibitors
- anti-TNF therapy
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
- autoinflammation
- congenital sideroblastic anemia with immunodeficiency
- fevers
- developmental delay (SIFD)
- tRNA
- TRNT1
- TNF inhibitors
- anti-TNF therapy
Introduction
Recent studies of rare monogenic systemic inflammatory diseases have illuminated the complexity of immune pathways through the discovery of patients who display overlapping features of autoinflammation, autoimmunity and immunodeficiency.1–5 Sideroblastic anaemia with immunodeficiency, fevers and developmental delay (SIFD) is one of these disorders caused by recessively inherited hypomorphic mutations in the housekeeping gene, TRNT1.6 7 To date, 23 patients have been reported, with significant clinical and immunological heterogeneity. At the severe end of the spectrum are patients with profound anaemia, immunodeficiency, fevers, developmental delay and metabolic aberrations leading to high morbidity and mortality. At the milder end of the spectrum are patients who present with retinitis pigmentosa and moderate haematological and immunological abnormalities. Although it has been hypothesised that mitochondrial dysfunction may play an important role in the pathogenesis of SIFD, the molecular mechanisms of the disease remain largely unknown.6–14
TRNT1 is a nuclear gene encoding the enzyme tRNA nucleotidyltransferase, CCA-adding, 1 (TRNT1) that is essential for maturation of cytosolic and mitochondrial tRNAs required for protein synthesis. The TRNT1 enzyme adds the 3-nt CCA sequence at the 3′ end of all precursor tRNAs, which is the site of the amino acid attachment catalysed by aminoacyl-tRNA synthetases.15 In addition, TRNT1 plays a role in surveillance of tRNA quality by selectively marking structurally unstable tRNAs for degradation.16 Surprisingly, only one gene has been identified in humans. Complete deficiency of TRNT1 is embryonic lethal.
Given the ubiquitous expression of TRNT1 and its critical function related to protein translation, it is expected that reduced expression of the enzyme will have a nearly global effect and lead to a complex phenotype. In this study, we aimed to explore the immunological features and possible mechanisms of inflammation and to explore the use of targeted therapy with cytokine inhibitors.
Methods
Patients and healthy controls
All the patients or their parents provided signed informed consent to participate in the study, which was approved by the NIDDK/NIAMS Institutional Review Board. Patients 2, 4, 5 and 6 were seen at the National Institutes of Health Clinical Center. For all patients, medical records were reviewed, including available outpatient material. The SI Materials and Methods describe the methods used for all the experiments.
Results
TRNT1 mutations
Independent of the published reports of TRNT1 disease-associated mutations, using WES, we identified biallelic loss-of-function (LOF) mutations in the common gene TRNT1 in two affected sisters from a consanguineous Saudi Arabian family (patients 1 and 2) and in a single affected child of mixed European ancestry (patient 3). All of them were referred to our clinic for evaluation of undiagnosed recurrent fevers (figure 1A,B). Patients 1 and 2 were homozygous for the p.His215Arg mutation, whereas patient 3 was compound heterozygous for the p.Asp163Val and p.Ile223Thr mutations. Subsequently, we identified six additional Caucasian patients, and here we report a total of nine patients and eight disease-causing mutations, three of which have not been previously associated with SIFD (p.Thr110Ile, p.Asp163Val and p.His215Arg) (figure 1A, online supplementary table 1). TRNT1 pathogenic variants in patients 4, 6 and 7 were also found by WES, while patients 5, 8 and 9 underwent targeted gene sequencing. Two unrelated patients 4 and 5 shared the same genotype and similar clinical and immunological features (see online supplementary tables 2 and 4). All TRNT1 pathogenic variants are either novel or found at a very low frequency in the general population (see online supplementary table 1). All missense disease variants affect evolutionarily conserved amino acid residues (figure 1C) and were computationally predicted to disrupt protein function. Four variants, Arg99Trp, Thr110Ile, Asp128Gly and Asp163Val, affect residues in the active-site domains that are close to the TRNT1 catalytic centre (figure 1D).
Supplemental material

Biallelic mutations in TRNT1 in the NHGRI cohort of patients. (A) Pedigrees of nine patients with SIFD. (B) Schematic representation of the exome data filtering approach leading to the identification of TRNT1 as the unique common gene in the first two families. (C) Evolutionary conservation of SIFD-associated mutations in this cohort. (D) In silico modelling of TRNT1 mutations based on the crystal structure of human TRNT1 (1ou5) (NP_001289875). Residues Arg99, Asp163, Thr110 and Asp128 are located within the head domain of TRNT1 and close to the catalytic residues Asp77, Asp79 and Glu121. Residues His215 and Ile223 are located in the neck domain. Lys416 and Ser418 are in the tail domain of the enzyme beyond the resolved crystal structure. (The PyMOL Molecular Graphics System, Schrödinger). SIFD, sideroblastic anaemia with immunodeficiency, fevers and developmental delay.
Clinical manifestations
Patients in this cohort presented with a spectrum of chronic disease manifestations including sideroblastic anaemia (SA), microcytic anaemia, variable degrees of immunodeficiency, vision and sensorineural hearing loss, developmental delay and failure to thrive. Detailed clinical characteristics are provided in figure 2 and online supplementary tables 2 and 3. Severe bouts of inflammation appeared as early as in the first 4–6 weeks of life in 7/9 patients, with patients 3, 4 and 5 having evidence for persistent systemic inflammation between flares. Although the duration of fever in SIFD typically lasts for 5–7 days, in patient 5, fevers progressively increased in length, lasting up to 2 weeks. Mucocutaneous and musculoskeletal manifestations occurred frequently, typically in association with fever, and included oral ulcers, skin cellulitis, dactylitis and arthralgia or arthritis of medium and small joints. As a result, patients are often referred to paediatric rheumatologists. Three patients experienced recurrent tender subcutaneous nodules. Skin biopsy from patient 7 revealed septal panniculitis and neutrophilic infiltration extending to the fibrous septa and adipose tissue, findings consistent with erythema nodosum. Patient 5 suffered from extensive soft tissue masses that were histologically consistent with mononuclear myofasciitis. The need for blood transfusions was often increased during fevers. In two patients, bone marrow biopsy revealed signs of haemophagocytosis, more pronounced in patient 3 who fulfilled part of the criteria for acquired lymphohistiocytosis syndrome, including neurological involvement (see online supplementary table 2). Finally, two patients experienced unprovoked recurrent thromboembolic events and these haematological findings have not been previously reported in SIFD.

Clinical findings of the NHGRI cohort of patients with SIFD. (A) Iron staining of patient 2 bone marrow aspirate showing ring sideroblasts (that represented more than 50% of erythroid precursors) (black arrows) and increased iron staining. (B) Abnormal findings in the cerebrum of patient 3 by fluid-attenuated inversion recovery (FLAIR) MRI of the brain. The image shows bilateral cerebral atrophy (yellow arrows), enlarged ventricles (white arrows) and extensive leukomalacia (red arrow heads). (C) H&E staining of rectosigmoid colon biopsy in patient 3 showing acute focal inflammation. (D) Splenomegaly in patient 2. (E) Knee effusion in patient 5. (F) Phagocyte (arrow) on bone marrow aspirate smear of patient 4. (G) Neutrophil with toxic granules in peripheral blood smear of patient 4. SIFD, sideroblastic anaemia with immunodeficiency, fevers and developmental delay.
Patient 1 died of multiorgan failure at the age of 7 years during a sepsis-like culture-negative febrile episode. Patient 3, who had prominent neurological deficits and severe B cell immunodeficiency, died at the age of 3 years, 92 days post peripheral blood stem cell transplant (see online supplementary table 2). Patient 7, who underwent a splenectomy for transfusion-dependent anaemia, died at the age of 9 years from Staphylococcus aureus sepsis. Her sibling, patient 6, aged 26 years, has relatively mild disease compared with other patients in the cohort (see online supplementary table 3).
Immune system dysregulation
In this cohort, hypogammaglobulinaemia was detected in 7/9 patients. Persistent B cell lymphopenia was observed in four patients (see online supplementary tables 2–5). Flow cytometric analysis of bone marrow aspirate showed a significant increase in the percentage of immature B cells (CD10+/CD20−) and a marked decrease in mature CD10−/CD20+ B cells (see online supplementary figure 1), findings suggestive of abnormal B cell maturation as a probable cause of B cell lymphopenia.6 17
Supplemental material
T cell numbers were mostly normal or in the low to normal range. However, a decreased CD4/CD8 ratio was observed in two patients. Three patients had abnormal findings on lymphocyte proliferation assays (online supplementary tables 4 and 5). Other abnormalities included decreased numbers of monocytes and neutrophils, circulating bands and toxic granulosis of neutrophils and a higher numbers of double negative T cells in two patients (see online supplementary tables 2, 3 and 6).
In the majority of patients, natural killer (NK) cell numbers were in the low to normal range. Multicolour flow cytometry showed a trend for NK cell activation in the patients (n=3) compared with age-matched healthy controls (n=3), based on higher frequency of surface activating and cytotoxic receptors. The differences did not reach statistical significance, perhaps due to small sample size (see online supplementary figure 2A–C). Similarly, we observed a trend for decreased IFN-γ production in NK cells, despite preserved cytotoxicity, a phenomenon that has been described in chronic viral infections (see online supplementary figure 2D).18
All patients with hypogammaglobulinaemia (7/9) received IgG replacement therapy since diagnosis. Serious bacterial or viral infections remained infrequent and were likely a consequence of the underlying immunodeficiency and/or treatment with immunosuppressive therapy (see online supplementary tables 2 and 3).
Inflammatory signature
High acute-phase reactants, proinflammatory cytokines and chemokines were documented in two patients who had clinically active disease at the time of sampling. Serum cytokine profiling in patient 4 at two different time points (CRP levels of 200 mg/L and 133 mg/L each, normal <3 mg/L) showed high levels of IL-6, IL-12p40, IL-18, IFN-γ and the IFN-induced chemokines IP-10 and MIG (figure 3A). Postmortem analysis of plasma samples from patient 3 showed a similar cytokine profile, in particular high levels of IL-6, IL-18, IP-10 and MIG (figure 3B). The same patients had elevated levels of endogenous inhibitors of TNF, soluble TNF receptors 1 and 2, as further evidence of ongoing inflammation. Levels of anti-inflammatory cytokines were not significantly elevated. The assessment of cytokine production in stimulated whole blood cells or peripheral blood mononuclear cells (PBMCs) (with TLR-agonists, mitogens, TNF and IFNγ) from four patients showed no statistically significant differences compared with healthy control cells (data not shown).
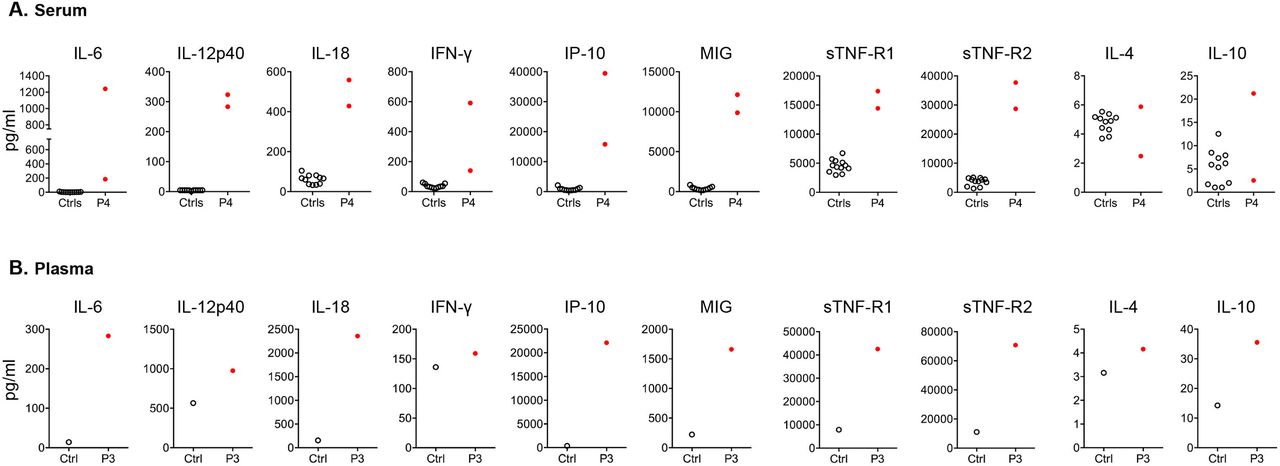
Inflammatory signature in patients with SIFD. (A) Serum cytokine and chemokine levels from patient 4 compared with paediatric healthy controls. For patient 4, each symbol represents measurements of serum samples collected during two independent flares (CRP levels 133 mg/L and 220 mg/L, respectively, normal values <3 mg/L). Forty-eight cytokines were measured by bead-based immunoassay as described in Material and Methods. Proinflammatory cytokines IL-6, IL-12p40, IL-18, IFN-γ and IFN-induced chemokines (IP10 and MIG) and levels of circulating soluble TNF-R1 (sTNF-R1) and TNF-R2 (sTNF-R2) were at least 10 SD higher in the patient compared with the average concentration of the 11 healthy controls. (B) Elevated levels of cytokines and chemokines, as shown for P4 in panel A, in postmortem plasma sample from patient 3 and a healthy control.
Electron microscopy
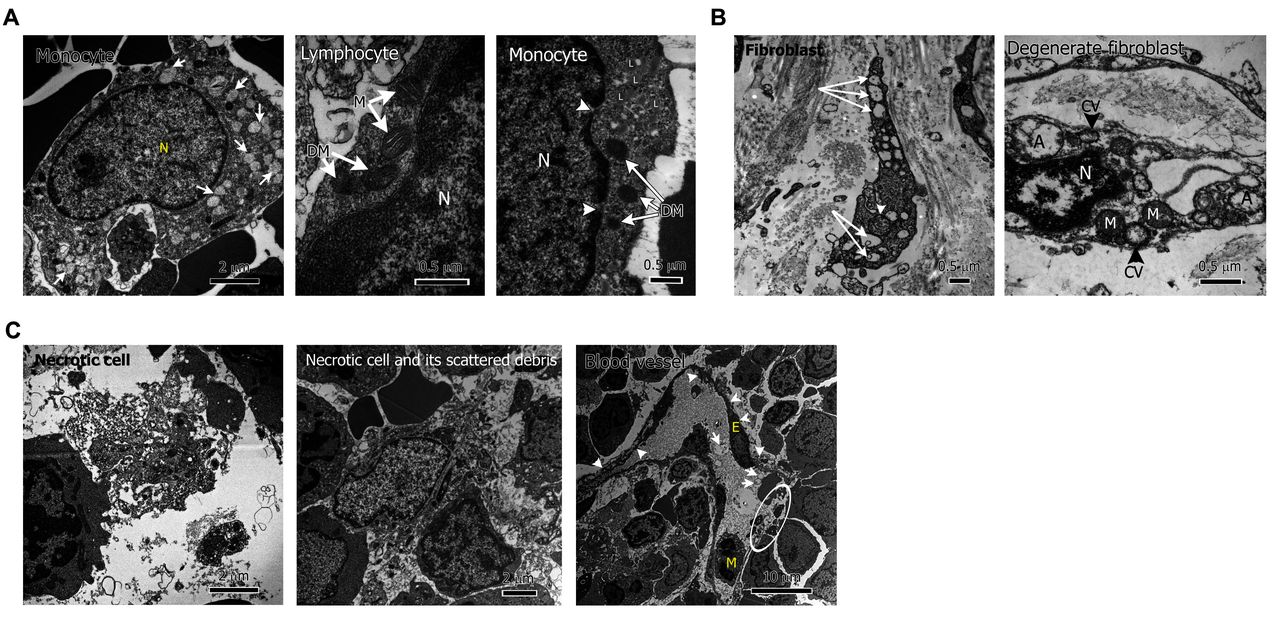
EM of bone marrow and skin biopsies showed a mixture of damaged, necrotic and normal-appearing cells. There was significant cellular degeneration with mitochondrial damage and numerous autophagosomes across all cell types (figure 4).

Ultrastructural findings of extensive cellular degeneration and necrosis. (A) Mitochondrial degeneration, from left to right: monocyte in patient 4 bone marrow (BM) with numerous degenerated mitochondria (white arrows); lymphocyte in patient 4 BM with coexistence of two normal mitochondria (M) and two abnormal, degenerate mitochondria (DM); monocyte in patient 6 BM with accumulation of lipid droplets (L), degenerate mitochondria (DM) (white arrows), nuclear envelope gaps (white arrowheads, N=nucleus). (B) Increased autophagosomes, from left to right: skin fibroblast in patient 6 with multiple autophagosomes (white arrows), degenerated mitochondrion (white arrowhead); degenerate fibroblast in patient 6 with pyknotic nucleus (N), extranuclear chromatin vesicles (CV), autophagosomes (A) and degenerate mitochondria (M). (C) Necrotic cells and debris in patient 4 BM (left and middle panel); blood vessel in P4 BM showing abnormal, degenerate endothelial cell (E), with thin, rough margins (white arrowheads), autophagosomes within serum (white arrows), vessel wall breakage and serum leakage (white circle) (M=monocyte).
Defect in maturation of tRNAs
We hypothesised that hypomorphic TRNT1 mutations will negatively affect the expression of mature cytosolic tRNAs in addition to previously reported mitochondrial tRNAs. We performed deep resequencing of mature tRNAs isolated from patient-derived fibroblasts (n=2) and healthy controls (n=3). Out of 227 tRNAs expressed in our dataset, 182 (80 %) were reduced in the patients compared with healthy controls. On average, the tRNA expression in the patients was 1.6-fold lower, a difference that was statistically significant (figure 5A).

Decreased mature tRNAs and proteostasis dysregulation in patients’ cells. (A) Defect in maturation of cytosolic tRNAs in patients’ fibroblasts. Briefly, tRNAs were purified from total RNA from patient (n=2) and healthy control (n=3) fibroblasts to prepare a small RNA library, followed by Illumina sequencing as outlined in Materials and Methods. Small RNA reads were aligned to the human genome and expression values for mature tRNAs, expressed in our dataset (n=227), were calculated as described in Materials and Methods. Left panel: plots show the density of the average expression of mature tRNAs in patients versus controls. Right panel: frequency of mature tRNAs in patients (n=2) relative to healthy controls (n=3). The x-axis of the histogram represents log2 ratio intervals and the y-axis represents the corresponding number of mature tRNAs in each ratio range. Statistics: one-sample Wilcoxon signed rank test. (B) Increased intracellular K48 ubiquitin levels and defects in autophagosome formation in mutant fibroblasts following stimulation with TNF and treatment with proteasome inhibitor MG132. Cultured fibroblasts from patients (n=2) and healthy controls (n=4) were detached from plates and treated with TNF with or without the proteasome inhibitor MG132 for 3 hours. Cells were lysed and immune-precipitated with antibodies against ubiquitin, and then blotted with specific antibody against K48-specific ubiquitin. (C) Bands in the immunoblots were quantified by Quantity One (Bio-Rad) and the ratio of LC3B-II/LC3 B-I is shown in bar graphs. TNF, tumour necrosis factor.
Increased reactive oxygen species (ROS) production in patients’ fibroblasts
Damaged mitochondria are a major source of intracellular ROS, which is a known trigger of inflammatory responses. Given the extensive mitochondrial abnormalities in patients’ cells (figure 4A), we measured ROS production in patients’ (n=3) and controls’ (n=2) fibroblasts. The number of live cells was slightly decreased in patients’ fibroblasts cultured for 72 hours, compared with controls (online supplementary figure 3A). The ROS production per live cells was higher in patients than controls (online supplementary figure 3B).
ROS accumulation is a known trigger of inflammasome-mediated pathways, including the NLRP3 inflammasome.19 20 We observed a higher production of IL-1β in the supernatant of stimulated monocyte-derived macrophages from patient 4 and in colon biopsy tissue from patient 5 by immunohistochemistry (online supplementary figure 4A, figure 6C). To further examine the effect of TRNT1 deficiency in myeloid cells, we knocked down TRNT1 in cultured human monocytic THP-1 cells. Small interfering RNA-mediated depletion of TRNT1 led to a significantly higher secretion of IL-1β in unstimulated and lipopolysaccharides (LPS) stimulated cells (online supplementary figure 4B,C). This effect was reversed in the presence of the NLRP3 inflammasome-specific inhibitor MCC950 (online supplementary figure 4C).
Deregulation of protein degradation pathways
Damaged mitochondria and proteins need to be efficiently removed by protein degradation pathways to prevent cellular stress and death. Given the evidence of necrosis and impaired protein synthesis in mutant cells, we investigated the function and efficiency of the ubiquitin-proteasome system (UPS) and the autophagy-lysosome system, which are responsible for removal and recycling of cell debris and damaged proteins.
Proteins that are targeted for degradation by UPS are marked by polyubiquitin lysine 48 (K48) chains. TNF-stimulated patients’ fibroblasts showed accumulation of high-molecular weight K48 Ub aggregates following treatment with the proteasome inhibitor MG132, compared with healthy control fibroblasts (figure 5B). Proteasome dysfunction activates autophagy.21 22 On induction of autophagy and formation of autophagosomes, LC3B-I protein converts to LC3B-II protein and the ratios of LC3B-II to LC3B-I and LC3B-II to actin are used to monitor autophagic activity. TNF-stimulated and MG132-treated patients’ fibroblasts displayed decreased LC3B-I to LC3B-II conversion compared with controls suggesting an impairment of autophagy (figure 5C). Under conditions of starvation that induces autophagy and chloroquine treatment that blocks the degradation of autophagosomes, both patient and control cells upregulated the autophagic activity as shown by increased LC3B-II levels (online supplementary figure 5A,B middle panels). However, mutant cells showed deficient clearance of the ubiquitin-scaffold protein p62 (sequestosome 1), which is a substrate for both autophagy and UPS (online supplementary figure 5A,B, top panels). This indicates a general inefficiency of TRNT1-deficient cells to upregulate protein clearance pathways under stress conditions.
Treatment with TNF inhibitors
Four patients have been treated with TNF inhibitors and showed steady improvement of clinical and laboratory parameters. Patient 5 was started on infliximab 12 years ago, patients 2 and 4 were started on etanercept following their molecular diagnosis, approximately 3 years ago, and patient 9 about 18 months ago. Anti-TNF therapy has been efficacious in suppressing fevers and normalising the inflammatory biomarkers (figure 6A,B). Immunohistochemical staining of colon tissue biopsies of patient 5 showed resolution of inflammatory infiltrates, primarily of neutrophils and macrophages, and reduction in staining for TNF and IL-1β (figure 6C). The frequency of blood transfusions in patients 2 and 4, both with a history of severe anaemia, decreased dramatically since the initiation of etanercept. Patient 9, who was entirely total parenteral nutrition (TPN) dependent, although on a suboptimal dose of etanercept, experienced a substantial improvement in enteropathy, which resulted in TPN wean and enteral feeding, and fewer blood transfusions.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Treatment with TNF inhibitors normalised the inflammatory signature in SIFD. (A) Serum CRP levels before and after treatment in patients 4 and 5. Asterisks indicate the initiation of therapy with etanercept for patient 4 (shown in red) and infliximab for patient 5 (shown in blue). (B) Normalisation of serum proinflammatory cytokines and soluble sCD14 levels, a marker of monocyte activation, in patient 4 following initiation of etanercept. (C) Patient 5 colon biopsy showing resolution of inflammation with anti-TNF therapy. Top row showing abnormal findings pretreatment: from left to right: H&E staining with cryptitis (blue long arrow), crypt abscesses (blue short arrow) and lymphoplasmatic infiltrates (white arrow); inflammatory infiltrates by neutrophils (MPO+); macrophages (KP1+) and CD8+ T cells in tissue immunohistochemistry; positive TNF and IL-1β immunohistochemistry staining. Bottom row: resolution of pretreatment abnormal findings following initiation of infliximab. (MPO: myeloperoxidase, KP1: macrosialin). CRP, C reactive protein; IL, interleukin; SIFD, sideroblastic anaemia with immunodeficiency, fevers and developmental delay; TNF, tumour necrosis factor.
We analysed the gene expression profile in whole blood samples of patient 4 before and after treatment with etanercept. We identified a set of 234 differentially expressed genes (DEGs) before treatment, compared with three age-matched healthy controls, that changed expression by at least two-fold following treatment with etanercept (online supplementary figure 6A). Pathway analysis assigned these DEGs into 52 statistically significant canonical pathways (online supplementary table 7 and supplementary figure 6B). They include pathways that are known to be regulated by TNF, such as leucocyte adhesion and trafficking, and endothelium activation.
Zebrafish model
CRISPR-mediated mutagenesis in zebrafish with complete LOF mutations recapitulated several phenotypes observed in patients with SIFD and confirmed the critical role of TRNT1 in development (online supplementary figures 7 and 8). The trnt1 −/− (null) zebrafish develop normally for the first 4 days as they can rely on the maternal sources of trnt1 present in the fertilised egg, but they all die of multisystem defects by nine days post fertilization (dpf) (online supplementary figure 7A).
Discussion
SIFD is a distinct autoinflammatory disease caused by mutations in the constitutively expressed enzyme, TRNT1, with a critical role in tRNA biogenesis. With the exception for SIFD and mevalonate kinase deficiency, other autoinflammatory diseases are caused by mutations in genes that are preferentially expressed in haematopoietic cells. The full continuum of clinical manifestations associated with decreased TRNT1 function is still emerging.
In this report, we extend the mutational spectrum and phenotypic attributes of SIFD, with a special emphasis on the immune and inflammatory manifestations. Clinical variability has been observed in this cohort and previously reported cases.6–14 Although congenital SA is one of the hallmarks of the disease, in three of our patients with chronic microcytic anaemia, SA was appreciated following the molecular diagnosis of the disease and re-evaluation of the bone marrow. Two of the patients carried a diagnosis of CVID, and notably the two siblings from Saudi Arabia had no evidence of B cell immunodeficiency. A significant clinical variability was observed among the affected siblings (families 1 and 5), which suggests a role for other modifying genes.
A previous study in SIFD demonstrated a deleterious effect of the TRNT1 deficiency on maturation of mitochondrial tRNAs.8 This is the first report to show a more global reduction in the expression of mature cytosolic tRNAs. Substantial mitochondrial damage and mitochondrial dysfunction in SIFD has been reported previously and in this study.7 8 11 12 Consistent with this cellular phenotype, we observed a higher spontaneous production of ROS in cultured patient fibroblasts. We also showed that TNRT1-deficient macrophages had increased production of IL-1β that was NLRP3 dependent.
The maintenance of protein homeostasis is essential to preserve cell functionality and survival. Rapid adjustments in protein translation are critical for cell survival and adaptation to stress. We showed that under stress conditions, mutant patient cells fail to upregulate protein clearance pathways. Perturbations in proteostasis are known to elicit activation of the innate immune system.23
EM in an actively inflamed patient was striking for the presence of necrotic and necroptotic cells, vascular leakage and extensive intercellular debris. Studies of cell death and inflammatory response continue to expand at the cellular and molecular levels. Dying and necrotic cells release an array of DAMPs and proinflammatory cytokines, such as IL-6 and IL-1, that increase chemoattraction of phagocytes to the site of necrosis, causing activation of inflammatory cascades.24–26 TNF is a potent stimulator and, after binding to TNFR1, can either stimulate downstream proinflammatory and survival signalling, that is, NF-kB pathway, or trigger cell death pathways and further perpetuate the inflammatory response.26 27 It is also possible that the accumulation of unprocessed/misfolded proteins causes endoplasmatic reticulum (ER) stress that activates the signalling unfolded protein response (UPR) network in TRNT-1-deficient cells, making them susceptible to TNF production. Although TNF levels were low in peripheral blood or supernatants of stimulated PBMCs, TNF was present in lesional biopsies along with inflammatory infiltrates consistent primarily of neutrophils and macrophages. Further studies are needed to identify TNF producing cells in SIFD.
Most patients are treated symptomatically, including red blood cells (RBC) transfusions, IgG replacement therapy and steroids, while the disease mortality remains high (median survival 48 months).6 This is the first report describing the clinical benefits of anti-TNF therapy for patients with SIFD. The dramatic response to the treatment with TNF inhibitors argues that TNF-dependent mechanisms have a major role in the pathogenesis of inflammation in SIFD. Our data suggest that TNF inhibitors suppress proinflammatory cytokines in the tissue and blood and decrease tissue inflammatory infiltrates. Two patients in this cohort had inefficient clinical response to treatment with IL-1β inhibitors (patient 4 and patient 8), similar to a single case reported previously.6 This indicates that IL-1 is contributing but not a major cytokine driving inflammation in SIFD.
The substantial decrease in requirement for blood transfusions following treatment with TNF inhibitors is likely related to improvements in anaemia of chronic disease. IL-6 is known to induce production of hepcidin, which in turn inhibits the differentiation of erythroid cells and decreases iron absorption.28 However, a chronic microcytic anaemia due to congenital SA persists in SIFD. Although hematopoietic stem cell transplantation (HSCT) was successful in ameliorating fevers and anaemia in a single patient, it is associated with increased risk for other complications.6 It is likely that TNF inhibitors do not have a direct effect on other TNF-independent disease processes, such as protein synthesis and turnover, although additional studies are required.
Collectively, our findings highlight the complexity of immune responses arising from mutations in TRNT1 and connect dysregulation in protein homeostasis to inflammation. Most important, our study is the first to report a new therapy for patients with SIFD, which can be especially beneficial if initiated early in life.
Acknowledgments
We would like to thank all the patients and their families, and the healthy controls, for their enthusiastic support during this research study.
References
Footnotes
Handling editor Josef S Smolen
Contributors AG, QZ, DLK and IA conceived the study. DLS, AS, RS, LS, DB, SJ, SP, DK, WCO, AAS, AKO, RHB, AAG, JMP, HCS and KR provided clinical data and patient material. AG, HW, QZ, YWP, MSA-A, KY, ES, WLT, DY, KB, BC, WP, ND, JHE, JC, EFR and GG-C performed experiments. AG, SB, YZ, JEN, SMB, MB, BR, MMQ, SMH, RS, KRC, SDR, MG, MH, H-WS and IA performed data analysis. AG and IA wrote the first and last draft of the manuscript. DLK and EFR critically revised the manuscript. All authors approved the final version of the manuscript.
Funding This research was supported by the Intramural Research Programs of the National Human Genome Research Institute (NHGRI), National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), National Heart, Lung, and Blood Institute (NHLBI), National Institute of Allegy and Infectious Diseases (NIAID), National Cancer Institute (NCI), National Eye Institute (NEI), Institute National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) and the NIH Clinical Center.
Competing interests None declared.
Patient consent Obtained.
Ethics approval The present study was approved by by the NIDDK/NIAMS Institutional Review Board.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Data are available from the corresponding author upon request.