Article Text

Abstract
Objectives To assess the effect of a temporary interruption in subcutaneous (SC) abatacept on immunogenicity, safety and efficacy in patients with active rheumatoid arthritis despite methotrexate in a phase III trial.
Methods Following a 12-week open-label introduction (period I; intravenous abatacept loading dose and weekly fixed-dose SC abatacept 125 mg), patients were randomised 2:1 to double-blind SC placebo or SC abatacept for 12 weeks (period II). At the end of period II, patients receiving SC abatacept continued treatment and patients on placebo were reintroduced to SC abatacept (12-week open-label period III). The co-primary end points were ELISA-detected immunogenicity rate and safety at the end of period II. Efficacy was also monitored.
Results Of 167 patients entering period I, 72% qualified for period II; during periods II and III, three patients discontinued treatment. Mean (SD) disease duration was 6.6 (6.5) years and Disease Activity Score 28 was 4.8 (0.8). The primary end point was met, with a non-significant increase in immunogenicity upon withdrawal (7/73 placebo vs 0/38 abatacept in period II; p=0.119) which was reversed upon reintroduction of SC abatacept (2/73 vs 1/38, end period III). Safety was comparable regardless of withdrawal, with no unexpected events upon reintroduction. Two patients experienced reactions at the SC injection site. On withdrawal, patients experienced slight worsening in efficacy which improved following reintroduction.
Conclusions Overall immunogenicity to SC abatacept is low, consistent with intravenous abatacept, and is not significantly affected by a 3-month interruption and reintroduction. This stop–start schedule was well tolerated, with little impact on safety and efficacy. These are important considerations for the clinical use of SC abatacept.
ClinicalTrials gov Identifier NCT00533897
This paper is freely available online under the BMJ Journals unlocked scheme, see http://ard.bmj.com/info/unlocked.dtl
Statistics from Altmetric.com
Introduction
Patients with rheumatoid arthritis (RA) receive long-term treatment and sometimes require interruption of therapy—for example, in the event of surgery or side effects. Such interruption in biological disease-modifying antirheumatic drugs (DMARDs) may lead to reduced drug concentration which may heighten the potential for drug-induced antibody responses.1 2 Immunogenicity can, in turn, reduce drug concentrations via antibody-mediated drug clearance, and reduce efficacy by preventing drug binding to the target. In addition, general immune-mediated toxicities such as infusion and subcutaneous (SC) injection site reactions may increase.1,–,3 This has been seen with the anti-tumour necrosis factor agent infliximab, for which serum sickness-like reactions have been observed at initiation and at reintroduction following extended withdrawal; such reactions were associated with increased immunogenicity, reduced serum concentrations and potential loss of efficacy.4,–,7
Experience with intravenous abatacept has demonstrated consistent safety and sustained efficacy, and a low immunogenicity rate that has no significant impact on pharmacokinetics, safety and efficacy.1 Theoretically, administration of abatacept via the SC route may have an increased risk for immunogenicity due to potential differences in antigen presentation.2 8 Fixed (125 mg/week) dosing of SC abatacept has so far demonstrated low immunogenicity rates and titres, and comparable drug exposure to intravenous abatacept.9 SC administration of abatacept is generally safe and well tolerated in patients with RA, with comparable efficacy to that seen with intravenous administration.9,–,11
This study assessed the effect of temporary withdrawal and reintroduction of 125 mg/week SC abatacept on immunogenicity, safety and efficacy in patients with RA.
Methods
Patients
Patients in the ALLOW (Evaluation of Abatacept Administered SubcutaneousLy in AduLts With Active RheumatOid Arthritis: Impact of Withdrawal and Reintroduction on Immunogenicity, Efficacy and Safety) trial met the criteria of the American Rheumatism Association (1987) for RA diagnosis and the American College of Rheumatology (1991) functional classes I, II or III. Patients had to have a Disease Activity Score 28 (DAS28; C reactive protein (CRP)) of 3.2–5.1 and received methotrexate (MTX) for ≥3 months prior to treatment.
Patients were excluded if they had serious acute or severe chronic/recurrent bacterial infection, or evidence of latent bacterial or viral infection, or were previously treated with rituximab, abatacept, live vaccine (≤3 months of first study dose or planned during study), leflunomide (≤1 year of screening) or any non-approved investigational biological agent.
Trial design
This multinational study consisted of three periods followed by a long-term extension (LTE) (figure 1). The study was conducted in accordance with the Declaration of Helsinki and was consistent with International Conference on Harmonization of Good Clinical Practice.12

Study design. *Most frequent reason for not being treated was no longer meeting entry criteria. †Patients who discontinued during period II (Withdrawal phase) due to lack of efficacy could enter period III (Reintroduction phase) prematurely. IV, intravenous; LTE, long-term extension; MTX, methotrexate; SC, subcutaneous.
Period I (Introduction) was an open-label (OL) 12-week period ending on day 85. On day 1, patients received a single intravenous dose of abatacept (∼10 mg/kg based on weight range; the ‘loading dose’), followed 30 min later by SC abatacept 125 mg. The single intravenous loading dose was given to ensure target therapeutic trough concentrations were achieved as quickly as possible. Thereafter, SC abatacept 125 mg was delivered at weekly intervals for the remainder of the 12-week period. Disease activity was assessed at day 78. Patients who had achieved an improvement in DAS28 (reduction of ≥0.6 from baseline; one times the measurement error and statistically considered a change) were randomised into period II; patients with DAS28 reduction <0.6 could directly enter the LTE.
Period II (Withdrawal) was a 12-week double-blind, placebo-controlled period ending on day 169, in which patients were randomised 2:1 to SC placebo or SC abatacept 125 mg/week. Patients could prematurely enter period III if RA symptoms persisted or worsened after symptomatic treatment in this period. This 12-week withdrawal period was determined to be sufficient for assessing development of immunogenicity because, at this time, serum concentrations of abatacept would be too low to interfere with the assay, and any immunosuppressive effect of abatacept that may interfere with immunogenicity would have ceased.
Period III (Reintroduction) was an OL 12-week period ending on day 253. To enter period III, patients had to have been treated in (but not necessarily completed) period II. In all patients regardless of randomisation group, OL SC abatacept 125 mg was self-administered weekly throughout period III. Patients who received SC placebo during period II were randomised (1:1) to receive either an intravenous placebo or intravenous abatacept loading dose (approximately 30 min before SC abatacept dosing at the beginning of period III (day 169)) to determine whether the administration of the intravenous abatacept loading dose affected immunogenicity upon reintroduction. Patients randomised to SC abatacept during period II received intravenous placebo loading on day 169 to maintain blinding.
A 4-week washout period was required for azathioprine, gold, etanercept or anakinra and an 8-week washout period was required for adalimumab or infliximab. Patients receiving sulfasalazine, chloroquine or hydroxychloroquine had to discontinue at screening.
Throughout the trial, low-dose (≤10 mg/day prednisone equivalent) oral corticosteroids were permitted but had to be stable for 25 of the 28 days before entry. Additionally, up to two high-dose corticosteroid courses were permitted during period II and one course during periods I and III (excluding 28 days before disease activity assessment at day 78 in period I). Biological and DMARD therapies other than MTX were not permitted. The MTX dose had to be stable (at least 10 mg/week) prior to study entry; increases in MTX dose were not permitted during the study, although decreases were allowed for toxicity.
Immunogenicity assessments
Blood samples were collected prior to intravenous infusions on days 1 and 169 and prior to SC injections on days 57, 78, 85, 113, 141, 197, 225 and 253. For the primary measure of immunogenicity, two validated ELISAs (Prevalere Life Sciences Inc., Whitesboro, NY, USA) were used; one determined the presence of antibodies to whole molecule abatacept (including both the CTLA4 and IgG portions) and one detected the presence of antibodies to the CTLA4 ‘tip’ portion alone (CTLA4-T). The primary end point of the study was the immunogenicity rate, expressed as the proportion of patients with an immunogenic response (ELISA) on day 169. In addition to ELISA, an electrochemiluminescence (ECL) immunoassay (Meso-Scale Discovery, Gaithersburg, Maryland, USA), which is more sensitive than ELISA, was used as a secondary assay and evaluated throughout. ELISA and ECL samples found positive in screening were verified by a second confirmatory assay and titre determination. Pharmacokinetic assessments were also made in this study and will be presented in a separate report.
Safety assessments
Adverse events (AEs) and serious adverse events (SAEs) were classified using the Medical Dictionary for Regulatory Activities (Version 12.1). Injection and infusion reactions were pre-specified and classified as SC injection site reactions (AEs at the site of SC injection) and acute infusion AEs (occurring within 1 h of the start of intravenous infusion). Autoimmune events were pre-specified based on the potential association with biological treatment.
Efficacy assessments
Efficacy assessments were secondary objectives; DAS28 and the Health Assessment Questionnaire-Disability Index (HAQ-DI) were evaluated at each visit. The proportions of patients achieving low disease activity state (LDAS; DAS28 ≤3.2) and DAS28-defined remission (DAS28 <2.6) were determined.
Statistical methods
The sample size of 105 patients, allocated 2:1 to receive SC placebo and SC abatacept during period II, was calculated to yield approximately 90% power to detect an absolute difference of 30% in the immunogenicity rate between period II treatment groups at the 5% significance level; this assumes an immunogenicity rate of 5% with SC abatacept at the end of period II and an overall discontinuation rate of 5% during period II.
Immunogenicity was analysed for patients with serum samples available at the visits of interest (as-observed) in period I (all patients treated during period I), period II (all patients randomised and treated in period II) and period III (all patients treated in period II and subsequently treated in period III). Immunogenicity rates are summarised over time using point estimates and within-group 95% CI, with 95% CI for between-group differences calculated at days 169 and 253. A continuity corrected χ2 test was used to compare the immunogenicity rate between groups on day 169 (p value evaluated at the 0.05 significance level).
Safety is reported for each period for all patients who received ≥1 dose of study drug during that period. Efficacy results are presented for patients treated in period II who had data available at the visit of interest (as-observed).
Results
Patient disposition and baseline demographics
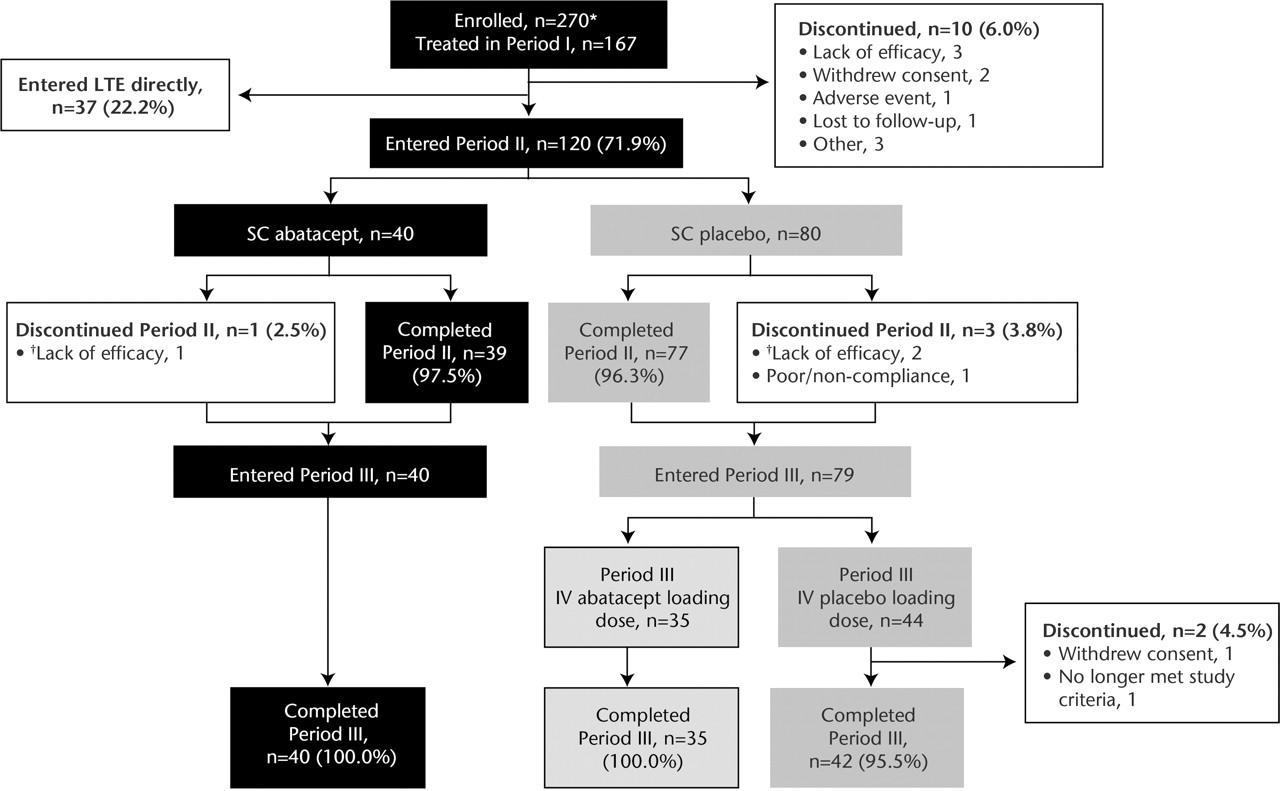
A total of 157/167 patients (94.0%) completed period I (Introduction); 120 (71.9%) qualified for period II (figure 2). In period II (Withdrawal), 40 and 80 patients were treated with SC abatacept and SC placebo, respectively. Forty and 79 patients entered period III (Reintroduction), with 40 and 77 completing period III.

Patient disposition. *Most frequent reason for not being treated was no longer meeting entry criteria. †Patients discontinued period II (Withdrawal phase) early due to disease flare and directly entered period III (Reintroduction phase). IV, intravenous; LTE, long-term extension; SC, subcutaneous.
For patients who were treated during period II, baseline disease characteristics were consistent with mild-to-moderate active inflammatory disease and were similar between treatment groups (table 1). All patients had received prior MTX treatment and >90% had not received biological agents.
Demographic and clinical characteristics at baseline* for patients treated in period II (Withdrawal phase)
In period I, 49.7% of patients received corticosteroids (oral and/or injectable); the mean (SD) oral dose was 3.1 (3.8) mg. Five patients (3.0%) received symptomatic treatment with high-dose corticosteroids in period I (oral (≥40 mg/day), intramuscular or intravenous). In period II, 57.5% of patients treated with SC abatacept and 55.0% of those given SC placebo received corticosteroids (mean (SD) oral doses 4.2 (5.0) vs 3.8 (4.6) mg). One patient (1.3%) in the SC placebo group received high-dose corticosteroids. During period III, 55.0% of patients treated with SC abatacept and 54.4% of those given SC placebo received corticosteroids (mean (SD) oral doses 3.6 (4.4) vs 3.4 (4.1) mg). One patient (2.5%) in the SC abatacept group received high-dose corticosteroids. During the course of the trial no patient received high-dose corticosteroids for ≥28 days.
Immunogenicity
End of period II (Withdrawal)
The immunogenicity rate (ELISA) at the end of period II (day 169) was low with a positive response in 0% (0/38) and 9.6% (7/73) of patients in the SC abatacept and SC placebo groups, respectively (estimate of difference 9.59 (95% CI 0.83 to 18.34), p=0.119; table 2). Of the 7/73 patients with a positive response in the SC placebo group at the end of period II, one patient was positive for anti-abatacept antibodies and six for anti-CTLA4-T antibodies.
Immunogenicity summary
Period III (Reintroduction)
Of the seven patients who were positive for antibodies at the end of period II, three patients remained positive after drug reintroduction; one was negative by the end of period III, one (period II SC placebo group) had persistent antibodies throughout period III and one had persistent positivity at all post-treatment assessments following discontinuation. At the last visit of period III (day 253), 1/38 patients treated with SC abatacept during period II and 2/73 patients treated with SC placebo during period II had positive responses (table 2). For two of these patients (one from each period II treatment group), antibodies were detected only at this assessment.
Immunogenicity following reintroduction of SC abatacept for patients randomised to SC placebo in period II was similar regardless of whether they received intravenous abatacept or intravenous placebo loading at the start of the reintroduction period (0/32 and 2/41 (both anti-CTLA4-T), respectively, at day 253; estimate of difference 4.88% (95% CI –4.50% to 14.25%)).
Immunogenicity by ECL
Immunogenicity assessed by this secondary assay was consistent with the ELISA observations (data not shown).
Safety
Overall, 49.1% of patients experienced an AE during period I. The frequency of AEs remained consistent during periods II and III and was similar between groups (table 3). The majority of AEs were mild or moderate. Throughout the study, SAEs were reported in six patients: three in period I (cholelithiasis, extreme fatigue, cellulitis and pulmonary embolism), two in period II (osteoarthritis of the right hip, and rectal fissure, laceration, pelvic fracture (motorcycle accident)) and one in period III (severe back pain, urinary retention and benign prostatic hypertrophy). The patient with pulmonary embolism died as a result of the event; all other SAEs resolved.
Safety summary
The frequency of infection was similar between groups in periods II and III (table 3). The most common infections (occurring in ≥5% of patients) were vaginal infection during period II and urinary tract infection, upper respiratory tract infection, laryngitis and flu during period III. Only two infections were severe (cellulitis and gastroenteritis salmonella), both of which occurred in period I. The cellulitis event was considered a serious infection and led to discontinuation.
Reactions at the SC injection site were rare and mild (table 3), with only two events reported (haematoma and pruritus in period I); no injection site reactions were reported upon drug reintroduction. Four patients (2.4%) in period I experienced hypertension of mild or moderate intensity within 24 hours of SC injection. One further moderate case was reported in period II in a patient withdrawn from SC abatacept. No anaphylactic reactions were reported.
Clinical efficacy
For patients treated in period II, mean (SD) DAS28 at baseline (day 1) was 4.80 (0.83). At the end of period I significant reductions from baseline in DAS28 were observed (figure 3A). For patients who continued to receive SC abatacept throughout period II (withdrawal), mean reductions were sustained throughout period II and remained stable during period III (figure 3A). For patients who had SC abatacept withdrawn in period II, DAS28 worsened slightly during withdrawal (figure 3A) and improved following reintroduction of SC abatacept and, by the end of period III (day 253), was comparable to patients maintained on SC abatacept (figure 3A). Mean DAS28 scores by the end of period III were similar, irrespective of whether or not patients received intravenous abatacept loading at reintroduction. High proportions of patients achieved DAS28-defined LDAS and remission by the end of period III, regardless of whether they were withdrawn from SC abatacept (figure 3B).
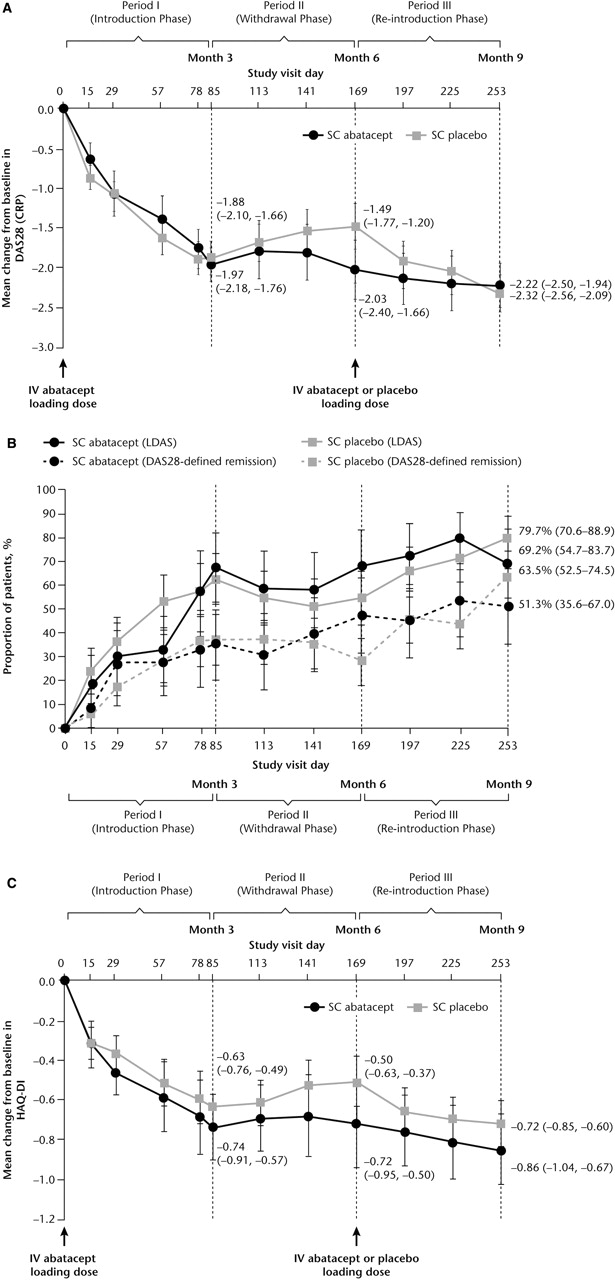
{kind=link}
{kind=link}
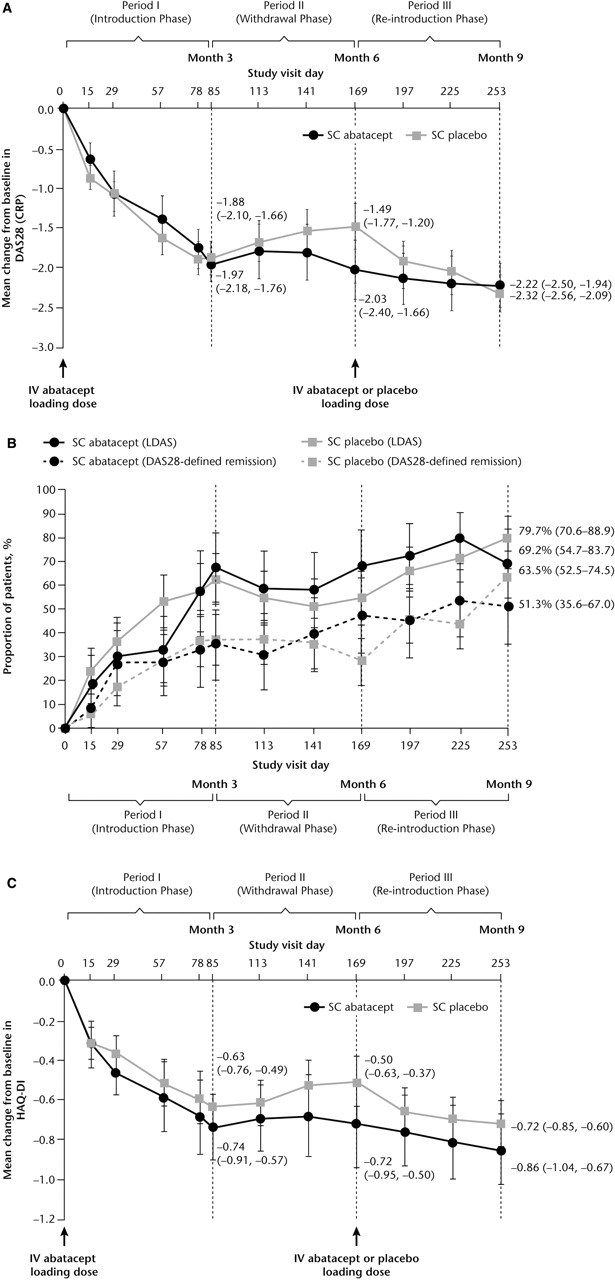{kind=link}
Clinical efficacy. (A) Mean change in DAS28 (CRP) over time by period II (Withdrawal phase) treatment group. (B) Proportion of patients achieving LDAS and DAS28-defined remission by period II (Withdrawal phase) treatment group. (C) Mean change in HAQ-DI score over time by period II (Withdrawal phase) treatment group. Data are as-observed for all patients who received ≥1 dose of study drug during period II. Error bars represent 95% CI. CRP, C reactive protein; DAS28, Disease Activity Score 28; HAQ-DI, Health Assessment Questionnaire-Disability Index; LDAS, low disease activity state; SC, subcutaneous.
Patients treated during period II had a mean (SD) HAQ-DI of 1.30 (0.66) at baseline. For patients maintained on SC abatacept during period II, mean reductions from baseline seen in period I were maintained through periods II and III (figure 3C). For patients withdrawn from SC abatacept, mean reductions in HAQ-DI seen in period I deteriorated slightly following withdrawal and improved upon reintroduction.
Impact of immunogenicity on efficacy and safety
Based on the seven patients treated with SC placebo who were seropositive (ELISA) at the end of period II, the presence of antibodies did not affect the safety or efficacy of SC abatacept. Following reintroduction, all but one of these patients demonstrated an efficacy response at the end of period III based on DAS28 and/or HAQ-DI. None of these seven patients experienced SAEs or severe or drug-related AEs after reintroduction, whether SC abatacept was reintroduced with or without intravenous abatacept loading.
Discussion
In clinical practice, patients may experience periods when treatment is temporarily withdrawn and reintroduced. Given the risk of increased immunogenicity and the potential impact on safety and efficacy resulting from treatment withdrawal and reintroduction, evaluating these outcomes is important.
The present study showed that, following a response to SC abatacept, 3-month interruption followed by reintroduction of treatment was well tolerated without a significant impact on immunogenicity. Following withdrawal of SC abatacept, immunogenicity was observed in approximately 10% of patients, with only one showing persistent seropositivity. The increase in immunogenicity for patients withdrawn from treatment was not significantly different from that of patients kept on treatment. After reintroduction of abatacept, <3% of patients demonstrated immunogenicity with only one case of persistent seropositivity. Immunogenicity was low regardless of whether intravenous abatacept or intravenous placebo was administered at reintroduction. Immunogenicity also appeared to have little effect on safety and efficacy, as shown in patients in whom treatment was withdrawn and subsequently reintroduced. Importantly, these data also demonstrate that immunogenicity with SC abatacept is consistent with intravenous administration.1 11
The low immunogenicity rates observed with SC abatacept mirror those observed in other SC abatacept studies. In a phase II study9 assessing weight-tiered versus fixed SC dosing over 4 months, only one of 51 patients demonstrated immunogenicity. This was transient and the patient was seronegative at study completion. Results from subsequent phase III studies have demonstrated low rates of immunogenicity. In a 4-month OL phase III study of SC abatacept with or without MTX, only four cases of transient immunogenicity were observed.10 Immunogenicity observed with SC abatacept9 10 is at the lower end of that seen with other biological agents. Immunogenicity rates of between 17% (21 patients) and 20% (46 patients) over 28 weeks have been seen with SC adalimumab,13 14 immunogenicity rates of approximately 6% (12 patients) over 24 weeks have been reported with SC etanercept15 and rates of 44% (33 patients) over 24 weeks have been found with intravenous infliximab.6
The safety of SC abatacept was consistent across periods during this study. Importantly, in patients in whom SC abatacept was withdrawn and reintroduced, the frequency of AEs did not increase and no unexpected events occurred compared with those who received continuous treatment. The incidence of infection with SC abatacept was consistent with intravenous administration, with the majority of events mild or moderate.16 17 There were no reports of autoimmune events or malignancies. The incidence of injection site reactions was low and all events were mild-to-moderate in intensity. Previous studies with SC abatacept have also reported a low incidence of injection site reactions,18 with fewer events seen with increasing treatment exposure.19
Patients who received continuous SC abatacept treatment had improvements in disease activity and physical function that were maintained over time, supporting the findings in patients with active RA who received SC abatacept with or without background MTX.19 Patients who withdrew SC abatacept treatment for 12 weeks experienced increased disease activity and slight worsening of physical function. Upon reintroduction, improvements in disease activity were noted within 1 month of re-initiation and, by the end of period III, disease activity was comparable with patients who received continuous SC abatacept. Similarly, physical function improved upon reintroduction of SC abatacept. These findings suggest that temporary short-term withdrawal of treatment has a minimal impact on sustained efficacy improvements seen in patients who respond to treatment. These are important findings, and provide some confidence for clinicians that interruption of SC abatacept treatment for up to 3 months is safe and well tolerated with a limited impact on efficacy.
Certain limitations of the study design should be considered. All patients received background immunosuppressive therapy with MTX, and caution should be used when extrapolating these conclusions to patients on monotherapy. However, a previous study determined that treatment with abatacept alone resulted in comparable immunogenicity to abatacept plus MTX, suggesting that the impact of background MTX on immunogenicity with SC abatacept is minimal.10 Patients were also permitted rescue therapy with high-dose steroids; however, the number of patients who received such rescue therapy is probably too low to influence the findings. Only one 12-week period of withdrawal was evaluated, so the impact of multiple treatment interruptions cannot be determined. However, 12 weeks is felt to be a representative length of withdrawal in clinical practice and is therefore clinically relevant. Furthermore, patients who did not experience any change in disease activity (DAS28 reduction <0.6 from baseline to day 78) were not randomised to the withdrawal period and entered the LTE directly. Such patients could be expected to be at a higher risk of immunogenicity, given the link between clinical response and immunogenicity seen with some biological agents.8 13 14 20 In this study a comparable immunogenicity profile was seen for these patients in the LTE to that seen for patients randomised and treated; this will be examined further in a future report.
It should be noted that the analyses presented here are based on as-observed data which are vulnerable to drop-outs. However, patient retention in this study was high. Lastly, the DAS28 results were based on CRP, which may yield higher responses than the DAS28 (erythrocyte sedimentation rate); however, CRP has been well validated as an outcome measure to assess disease activity21 and is increasingly used in clinical trials.
In summary, 3-month interruption with subsequent reintroduction of SC abatacept was associated with overall low immunogenicity, a finding consistent with that found with intravenous abatacept. Furthermore, this stop–start schedule was well tolerated and did not have a negative effect on efficacy. These findings are important considerations for the clinical use of SC abatacept.
Acknowledgments
The authors thank Eve Guichard, Medicus International, for editorial support.
References
Footnotes
-
Funding This study was sponsored and editorial support funded by Bristol-Myers Squibb.
-
Competing interests JK has received research grants from Bristol-Myers Squibb, Pfizer, Abbott, Vertex, Astra Zeneca, Chelsea, Lilly, Roche, Centocor, Merck, Amgen, UCB, Aventis and has participated in speakers' bureaux for Amgen, Bristol-Myers Squibb, Novartis Pharmaceuticals Corporation and UCB. GG has received research grants from Pfizer, Genentech, Roche, Forest, Cephalon, Regeneron, Centocor, Bristol-Myers Squibb and New England Research Associates, has participated in a speakers' bureau for Bristol-Myers Squibb and has received consulting fees from Bristol-Myers Squibb. IS has received consulting fees and honoraria from, and has participated in a speakers' bureau for, Bristol-Myers Squibb. MR and IL have no competing interests to declare. CL has received clinical research grants from, and has participated in speakers' bureaux for, Bristol-Myers Squibb. SG, RP, ID and MP are full-time employees of, and stockholders in, Bristol-Myers Squibb.
-
Ethics approval The study was approved by Institutional Review Board/Independent Ethics Committee and informed consent was given by patients.
-
Provenance and peer review Not commissioned; externally peer reviewed.