Article Text

Statistics from Altmetric.com
To achieve early and complete reperfusion of the myocardium in acute coronary syndromes is the daily challenge for every physician in clinical cardiology. However, restoration of epicardial blood flow by thrombolysis, primary angioplasty or bypass surgery does not necessarily imply complete reperfusion, even if the target stenosis is adequately removed or bypassed. The amount of microvascular integrity may limit reperfusion to the previously ischaemic tissue despite complete restoration of epicardial vessel diameters.
A 74 year old man with acute distress is admitted to the emergency room because of acute onset of severe, substernal, crushing chest pain two hours ago. He has never suffered from similar symptoms before. The ECG shows ST segment elevation in leads I, aVL, V2–V4. After aspirin and heparin, the patient is immediately transferred to the catheterisation laboratory. Coronary angiography confirms a thrombotic occlusion of the proximal left anterior descending artery. The guide wire easily crosses the occlusion. After coronary artery balloon dilatation and stent implantation the epicardial artery appears to have gained sufficient luminal diameter. However, the contrast medium is only slowly conveyed to the distal artery and not adequately washed out. Even the final angiogram after glyceryl trinitrate in different projections shows no satisfactory flow albeit no visible flow limiting obstacles, such as coronary artery dissection or recurrent thrombus formation. The battle, undertaken to restore myocardial blood supply, seems to be lost and won, finally leading to compromised tissue perfusion despite a successful restoration of patency to the epicardial blood vessel.
A bolus of abciximab, followed by a continuous infusion, is initiated. ST segment elevations resolve only slightly during the next hours; the patient requires prolonged intensive medical care because of recurrent pulmonary oedema. Finally, the patient is stabilised on cardiovascular medication including an angiotensin converting enzyme (ACE) inhibitor, oxygen, and aspirin. The echocardiography shows a large akinetic segment of the anteroseptal wall with an apical aneurysm, and a global ejection fraction of 30%.
Angiographic no-reflow, as described above, may be one of the clinically most obvious manifestations of microvascular damage. The following discussion will set forward that the so-called “no-reflow” phenomenon is far more frequent, and has important prognostic implications. Combining results from basic science and clinical research, we try to elucidate major mechanisms of no-reflow, methods of assessing microvascular dysfunction, and its significance in the clinical setting.
WHAT IS KNOWN ABOUT NO-REFLOW FROM ANIMAL STUDIES?
Anatomical no-reflow
In experimental models of myocardial infarction, injection of various dyes to the circulation has been traditionally used to stain perfused tissue. As shown in fig 1, a substantial part of the myocardium, after being subjected to proximal coronary occlusion and reperfusion of the epicardial artery, may remain unstained after injection of monastral blue or thioflavin S (a fluorescent vital stain for endothelium). The resulting visual perfusion defects represent the so-called “anatomical” no-reflow. Systematic investigations in the canine model of myocardial infarction demonstrated a homogenous distribution of thioflavin S after 40 minutes of ischaemia with subsequent reperfusion. However, after 90 minutes, areas of no-reflow were identified as zones not stained by the fluorescent dye,1 indicating that a certain duration of ischaemia is necessary for the development of no-reflow. In the canine model, these zones of no-reflow first appeared in the subendocardium.

Anatomical no-reflow, visualised in a rabbit model of coronary occlusion and reperfusion. Monastral blue (A) and thioflavin S (B, photography under ultraviolet light, Minolta Y48 barrier filter) were injected into the left atrium after 120 minutes of coronary artery occlusion and 120 minutes of reperfusion. Both dyes leave a substantial part of the left ventricle unstained, indicating areas of no or low perfusion. A close look at the slices demonstrates that (although closely correlating) the area not stained by monastral blue is slightly larger than the non-fluorescent area, reflecting the ability of the two dyes to penetrate into areas of hypoperfusion.
Ultrastructural changes
Electron microscopy of these areas of no-reflow revealed swollen intraluminal endothelial protrusions and membrane bound intraluminal bodies, which appeared to be free floating and often seemed to obstruct the capillary lumen. Endothelial cells showed decreased numbers of pinocytotic vesicles and nuclear chromatin margination. After 20 minutes of reperfusion, capillaries contained tightly packed erythrocytes, endothelial gaps, sometimes plugged by platelet and fibrin thrombi, with numerous extravascular red blood cells. Occasionally, capillaries seemed to be compressed by subsarcolemmal blebs of neighbouring swollen myocytes.1 Ultrastructural investigations after different durations (20–180 minutes) of occlusion in a dog model of coronary ligation and reperfusion demonstrated that microvascular damage (first observed after 60 minutes of occlusion) always lagged behind myocardial cell injury (first apparent after 20 minutes of ischaemia) and that microvascular alterations were always located in zones of irreversibly damaged myocytes.2 Areas not stained by the fluorescent dye were characterised by low regional myocardial blood flow (mean (SEM) 0.13 (0.03) ml/g/min to 0.26 (0.04) ml/g/min), as assessed by radioactive microspheres.3
Reperfusion injury and microvascular dysfunction
The term “reperfusion injury” has been coined to describe reperfusion related worsening or expansion of various forms of ischaemic cardiac damage, including: alterations of contractile performance; the arrhythmogenic threshold; potential conversion from reversible to irreversible cardiomyocyte injury; and vascular, in particular microvascular, dysfunction. Ambrosio and colleagues demonstrated a more than twofold increase in the area of no-reflow from 2 minutes to 3.5 hours of reperfusion. This increase was accompanied by a progressive decrease of regional myocardial blood flow within the risk area after initial restoration of regional myocardial flow.3 Thus, on the one hand no-reflow depends on the degree of the initial ischaemic damage, but on the other hand reperfusion injury at the microvascular level is a significant determinant of the final amount of no-reflow.
WHAT IS KNOWN FROM CLINICAL RESEARCH?
Angiographic no-reflow
Coronary angiography allows a semiquantitative grading of epicardial antegrade flow according to the thrombolysis in myocardial infarction (TIMI) flow grades. TIMI grade 0 refers to no distal antegrade vessel opacification by the contrast medium, grade 1 means minimal distal flow, grade 2 partial reperfusion, and grade 3 complete reperfusion. After thrombolysis or coronary angioplasty (PTCA) for acute myocardial infarction, TIMI 0–2 grade coronary flow is associated with more complications and worse left ventricular function compared with TIMI 3 grade flow, even without evidence for obstruction of the epicardial artery. In a retrospective review of 1919 elective and emergency percutaneous interventions by Piana and colleagues, “angiographic” no-reflow, defined as TIMI grade flow less than 3 without evidence for obstruction in the proximal artery, occurred in 2% of all interventions. But the incidence varied with the technique and indication for PTCA: the highest incidence of no-reflow was found after recanalisation for acute myocardial infarction (11.5%); the incidence was significantly higher for interventions in degenerated saphenous bypass grafts (4%); and for stenting or directional atherectomy interventions (3%).4 Recent studies demonstrated that angiographic no-reflow is a strong predictor of major cardiac complications, including congestive heart failure, malignant arrhythmias, and cardiac death after myocardial infarction.5
Myocardial contrast echocardiography
Myocardial contrast echocardiography (MCE) has profoundly extended our understanding of myocardial microvascular perfusion after acute myocardial infarction, and intravenous contrast application with bedside imaging is now feasible. Ito and colleagues showed that all patients with TIMI grade 2 flow after PTCA showed substantial no-reflow on MCE, defined as contrast defects after angioplasty of more than 25% of the risk zone (determined before recanalisation). But even with TIMI grade 3 flow, 16% of the patients showed no-reflow, and significant improvement of left ventricular function was only observed in the patients with reflow.6 Comparing microvascular perfusion and regional myocardial contractile recovery, Ragosta and associates concluded that microvascular integrity, as assessed by MCE, is closely related to myocyte viability.7 Sakuma and colleagues demonstrated that the size of the risk area, determined before reperfusion, as well as a low peak grey scale ratio on MCE one day after primary PTCA for acute myocardial infarction, strongly predicted major cardiac events within the next 22 months.8 Risk factors for the development of perfusion defects on MCE were not consistent in different studies, but most of them reported a higher incidence of no-reflow with longer elapsed time from onset of symptoms to reperfusion, older age, large anterior myocardial infarction, low admission blood pressure, and, interestingly, with the absence of pre-infarction angina, which might be interpreted as a clinical correlate of microvascular preconditioning.
Intracoronary Doppler flow velocity changes
Intracoronary Doppler derived flow velocity patterns showed characteristic profiles in patients with evidence for no-reflow on MCE, including early systolic retrograde flow and high diastolic deceleration rates, and a lower coronary flow reserve.9 Iwakura and colleagues showed that patients with occurrence of these Doppler flow characteristics immediately after PTCA had longer elapsed time from onset of symptoms to reperfusion and more Q waves on the surface ECG before reperfusion. Patients with delayed (within 10 minutes) systolic retrograde flow had a higher incidence of transient ST segment re-elevation after recanalisation.10 The delayed occurrence of these characteristics in a subgroup of patients might be a correlate of reperfusion associated progression of no-reflow.
TIMI frame count and myocardial blush grade
Other angiographic techniques, used for assessing microvascular integrity in patients with reperfused myocardial infarction, are the corrected TIMI frame count, defined as the number of angiographic frames required for the contrast medium to reach standardised distal landmarks of the coronary artery, and myocardial blush grade, as a semiquantitative description of myocardial contrast density on the final angiogram after reperfusion therapy.
Scintigraphic techniques
Visualisation of perfusion defects was also achieved by scintigraphic techniques. Intracoronary injection of macroaggregated 99mtechnetium albumin after successful percutaneous recanalisation demonstrated substantial perfusion defects. In addition, evidence for a progressive impairment of tissue perfusion during reperfusion was provided by 82rubidium positron emission in a canine model.
Magnetic resonance imaging
In magnetic resonance imaging (MRI), hypoenhancement 1–2 minutes after contrast injection is assumed to represent zones of no-reflow. A recent study validated the amount of hypoenhancement against anatomical no-reflow assessed by injection of thioflavin S and regional myocardial blood flow in a canine model of reperfused myocardial infarction.11 Assessing hypoenhancement after contrast injection, visualised in 44 patients 10 days after acute myocardial infarction, Wu and colleagues demonstrated significant prognostic implications of microvascular obstruction for clinical outcome, even after statistical correction for the predictive value of infarct size.12 Thus, contrast enhanced MRI might be a promising non-invasive future technique for the determination of microvascular obstruction with the opportunity of simultaneous estimation of infarct size (visible as hyperenhancement 5–10 minutes after contrast injection).
Resolution of ST segment elevation
The resolution of ST segment elevation after reperfusion therapy was shown to correlate with the amount of myocardial perfusion as visualised by MCE. The so-called ST segment elevation index refers to the sum of ST segment elevation (mV) in the different leads (excluding aVR), divided by the number of leads showing ST segment elevation. While patients without evidence of no-reflow showed a rapid reduction of the ST segment elevation index within the first 30 minutes after successful primary PTCA, ST segment elevation in patients with no-reflow resolved to a significantly lesser degree. Indeed, a (transient) re-elevation was observed in some of these patients.13
Biochemical markers
Serial measures of serum myoglobin, creatine kinase-MB or troponin I (or T) at baseline and 60 (or 90) minutes after reperfusion therapy have been a useful technique for the assessment of infarct related artery patency. A high ratio of the 60 minute value to the baseline value and a higher slope of increase in these markers over time are generally related to successful reopening of the epicardial occlusion, but may also reflect the completeness of microvascular tissue reperfusion. Thus, the amount of tissue reperfusion after reperfusion therapy might in part be estimated even without invasive procedures or sophisticated and expensive technical equipment.
POTENTIAL MECHANISMS OF NO-REFLOW
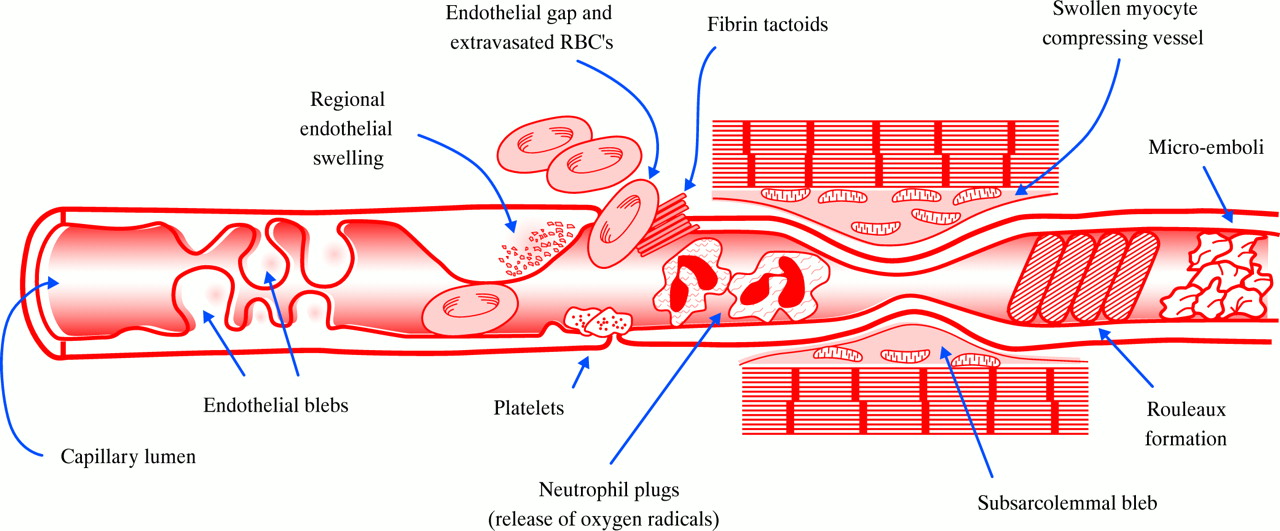
Although numerous potential explanations of the development of no-reflow have been put forward (fig 2), the significance of any single mechanism is not yet fully understood. In addition, the importance of these mechanisms might be different immediately after release of the coronary occlusion versus later during the time course of reperfusion. Mechanisms causing no-reflow in experimental mechanical coronary occlusion and reperfusion may differ from those of clinical myocardial infarction or observed during percutaneous coronary interventions (for a list of references refer to eHeart).
{kind=link}
{kind=link}
Schematic figure, summarising different mechanisms, involved in the development of no-reflow, and accompanying ultrastructural alterations of the microvascular bed (RBC, red blood cell).
Endothelial ischaemic damage and microvascular obstruction
The pronounced ultrastructural alterations of the capillary endothelium demonstrated in the canine model suggest that morphological features of the microvascular damage related to ischaemia may directly contribute to the no-reflow phenomenon. Localised areas of endothelial swelling and endothelial protrusions (also called “blebs”) were the most common findings.1 These protrusions may act to occlude the capillary lumen, and thus play a direct role in causing regional perfusion defects, probably most important directly after reopening of the coronary artery. Following reperfusion, capillaries showed tightly packed erythrocytes suggesting that some flow must have occurred initially into these regions after release of the coronary occlusion.
Leucocytes: plugging versus interaction with endothelium, platelets, and myocytes
The pronounced increase of anatomical no-reflow during reperfusion, observed in the experiments by Ambrosio and colleagues, was accompanied by striking neutrophil accumulation within no-reflow areas.3 Engler and associates demonstrated extensive leucocyte plugging in the microvasculature, that could not be washed out by crystalloid perfusion, after coronary occlusion and five hours of reperfusion. Leucocyte depletion or reperfusion with oxygenated perfluorochemicals led to a reduction of anatomical no-reflow in various studies. However, it should be emphasised that even in isolated saline perfused Langendorff hearts, an anatomical area of no-reflow can be visualised. Thus, leucocytes may exacerbate no-reflow, but do not appear to be a requirement for it to occur.
Even if capillary leucocyte trapping is prominent in the area of no-reflow, the effects of leucocytes are probably not solely confined to mechanical plugging, but may involve complex interactions with the endothelium, platelets, and perhaps with myocytes. Polymorphonuclear cells are able to release reactive oxygen metabolites, proteolytic enzymes, and lipooxygenase products (leukotrienes) that influence platelet and endothelial function. Endothelial cells can modulate leucocyte function by the expression of adhesion molecules—for example, intercellular adhesion molecule-1 (ICAM-1) or P-selectin—and by release of soluble factors including nitric oxide, prostacyclin, endothelin, and platelet activating factor. Platelets affect polymorphonuclear cell activation by release of thromboxane A2, platelet derived growth factor, serotonin, lipooxygenase products, proteases, and adenosine. However, various studies addressing these questions led to partly inconsistent and contradictory results, depending upon the specific model or species investigated. Clinical trials investigating the effect of neutrophil inhibition in myocardial infarction have been in general, negative.
Reactive oxygen species
The production of oxygen-free radicals peaks during the first 2–10 minutes of reperfusion after coronary artery occlusion. Sources of oxygen free radicals include the xanthine oxidase reaction, mitochondria, and polymorphonuclear cells. The superoxide dismutase pathway, normally responsible for the clearance of superoxide anions, may be altered after an ischaemic insult. Administration of superoxide dismutase in combination with catalase led to a notable reduction of no-reflow, and ultrastructural signs of endothelial injury in animal studies14; also free radical scavengers were shown to prevent functional vascular dysfunction after ischaemia and reperfusion.
Functional abnormalities of the vessels
Vascular dysfunction after ischemia and reperfusion in animal models is characterised by a decreased vasodilation in response to acetylcholine or platelet derived stimuli. Functional vascular alterations were shown to persist for several weeks. Interestingly, ischaemic preconditioning can prevent functional vascular abnormalities.15 Clinical observations confirmed a reduced coronary flow reserve after ischaemia and reperfusion. But even if impaired endothelial response to physiologic stimuli is not the primary cause of anatomical no-reflow, functional vascular abnormalities, especially long term alterations, may prevent adaptation of regional blood flow to situations with an increased oxygen or substrate demand.
Mechanical compression
Sudden myocardial cell swelling with prominent intracellular and interstitial oedema is one of the very early morphologic changes induced by reperfusion. As tissue oedema might compress the microvascular bed, the no-reflow phenomenon may in part be attributed to changes in total cross sectional vascular area. However, studies investigating the effect of increased serum osmolality to reduce tissue oedema did not show consistent results. It is likely that mechanical compression contributes in some situations, but is not the primary mechanism of no-reflow.
Coagulation, tissue factor
Ultrastructural investigations of no-reflow areas did not show direct evidence of a causal role of intravascular thrombus formation for the development of no-reflow in animal models of mechanical coronary artery occlusion and reperfusion. Some electron microscopy studies found rare microthrombi1; others did not find any evidence for activation of the coagulation pathway. Administration of acetylsalicylic acid, streptokinase or tissue plasminogen activator did not demonstrate any beneficial effects on microvascular integrity.
Recent observations suggest that tissue factor, a membrane bound glycoprotein that activates the extrinsic coagulation pathway (via activating factor VII) when exposed to flowing blood, contributes to the degree of no-reflow. When active site-blocked factor VII was administered during reperfusion, a pronounced reduction of no-reflow was observed,16 which might be of special interest in clinical acute coronary syndromes, associated with vascular and endothelial damage.
Microembolisation of atherosclerotic debris and thrombi
Unlike most animal models of mechanical coronary occlusion, the clinical setting probably involves microembolic events in a substantial number of cases. The transition of a stable atherosclerotic lesion to an unstable plaque, induced by inflammatory stimuli or by iatrogenic manipulation, includes the disruption of the fibrous cap with exposure of subendothelial matrix, which subsequently can lead to embolisation of lipid, matrix, endothelial cells or platelet thrombi to the distal vasculature.17
In unstable angina, elevation of serum troponin T is a very sensitive marker of myocardial injury, reflecting micronecrosis caused by distal coronary embolisation. The pronounced beneficial effects of glycoprotein IIb/IIIa receptor antagonists in the subgroup of patients with unstable angina and increased troponin T serum concentrations, evident in the CAPTURE (chimeric 7E3 antiplatelet therapy in unstable angina refractory to standard treatment) study, indicates a role of microembolisation followed by platelet activation in this subgroup.18 Cyclic flow variations in stenosed coronary arteries are regarded as the consequence of the formation of platelet aggregates and subsequent dislodgement of the embolus into the microvascular bed.
Elective angioplasty procedures are associated with a certain incidence of the no-reflow phenomenon.4 Depending on the technique, the incidence is quite variable. Patients who undergo percutaneous interventions for degenerated saphenous vein grafts have a high risk of periprocedural infarction. Mechanical devices, designed to trap embolic debris during angioplasty, showed that embolic material could be retrieved during PTCA, especially in angioplasty interventions in degenerated venous grafts, which is “smoking-gun evidence” for the concept of PTCA associated distal embolisation. Directional and rotational atherectomy have a substantially higher risk of no-reflow compared with balloon angioplasty. Thus, microemboli significantly contribute to the no-reflow phenomenon in the clinical situation, leading to micro- and potentially macro-infarcts.
TREATMENT OF NO-REFLOW: DOES IT MAKE SENSE?
Obviously, the prevention of microembolisation or its consequences should be beneficial in the clinical setting. Glycoprotein IIb/IIIa receptor blockers, that should reduce the detrimental sequelae of coronary embolisation in the microvascular bed, have demonstrated convincing effects on clinical outcome in unstable angina and after percutaneous intervention, in addition to prevention of reocclusion of the target lesion.
However, with respect to the “classical” no-reflow phenomenon in coronary occlusion and reperfusion, the question is more difficult to answer. To date, no study could demonstrate no-reflow zones preceding myocardial necrosis. In 312 biopsies of ischaemic canine myocardium after various times of coronary occlusion (20–180 minutes), no biopsy showed microvascular damage without myocardial cell injury. In addition, ultrastructural myocyte damage greatly preceded microvascular alterations.2 Even in the experiments of Ambrosio and colleagues, that showed a pronounced expansion of no-reflow during reperfusion, the area of hypoperfusion was always confined to the area of necrosis.3
As there is no proof of a contribution of classical no-reflow to myocardial necrosis, one might ask whether treatment that focuses on reduction of no-reflow in necrotic tissue makes sense. In theory, improvement of tissue perfusion, even in areas of irreversibly damaged cardiomyocytes, could have beneficial effects other than myocardial salvage. Improved tissue perfusion might impede infarct expansion, ventricular remodelling or aneurysmic ventricular dilation, and promote scar healing and the delivery of pharmacological agents to the myocardium. Furthermore, blood vessels preserved in an area that might have become a no-reflow zone could serve as a source of future collateral vessels.
The MRI study by Wu and colleagues demonstrated prognostic significance of no-reflow after acute myocardial infarction independent of its relation to infarct size. The presence of microvascular obstruction in this study was significantly correlated with fibrous scar formation, left ventricular remodelling after six months, and worse clinical outcome.12
TREATMENT OF NO-REFLOW: WHAT TREATMENTS WORK?
Treatment strategies to reduce no-reflow have achieved increasing recognition both in basic animal research and clinical studies. However, pharmacological interventions are not finally established except for the prevention of the detrimental effects of coronary microembolisation by special devices or therapies that reduce platelet activation. Table 1 summarises a selection of interventions that were shown to exhibit beneficial effects on no-reflow in animal models; table 2 focuses on treatment strategies used to reduce no-reflow in clinical situations. Pharmacological agents with vasodilating properties, such as adenosine, papaverine, and calcium antagonists were investigated in experimental and clinical conditions. The results from animal studies were controversial with respect to adenosine. Clinical studies mainly used angiographic no-reflow as an end point, which might not fully reflect microvascular perfusion. The beneficial effects of leucocyte depletion or oxygen radical scavenging in animal studies did not show convincing usefulness in the clinical setting. As demonstrated recently, nicorandil administered directly after the diagnosis of acute myocardial infarction was effective in both reducing the perfusion defect (MCE) and complications after infarction, accompanied by an increased recovery of left ventricular function.19 In another study, intracoronary verapamil was also shown to reduce perfusion defects and increase peak contrast intensity in MCE after primary PTCA for acute myocardial infarction.20
Interventions used in experimental animal models to reduce no-reflow
Interventions used in clinical situations to reduce no-reflow
In summary, the best treatment strategy for no-reflow has not yet been characterised, and future investigations are needed to evaluate the significance and contribution of different mechanisms responsible for no-reflow in order to provide optimal therapy.
No-reflow phenomenon: key points
-
Experimental no-reflow is characterised by microvascular dysfunction, evident as distinct areas of hypoperfusion, compromised resting myocardial blood flow, and functional vascular alterations after ischaemia and reperfusion
-
Clinical observations, using TIMI grading of coronary flow, MCE, scintigraphic, and MRI techniques show evidence for similar features of microvascular damage after acute myocardial infarction, and even after elective PTCA interventions
-
Coronary microembolisation is an important mechanism of no-reflow in acute coronary syndromes and PTCA procedures
-
Perfusion defects after acute myocardial infarction—for example, visualised by MCE—are closely related to lack of contractile recovery and irreversible myocyte damage
SUMMARY AND CONCLUSIONS
Both animal models of experimental myocardial infarction and clinical studies have provided evidence of impaired microvascular perfusion after reperfusion. Characteristics of no-reflow found in basic science investigations, such as distinct perfusion defects, progressive decrease of resting myocardial flow with ongoing reperfusion and functional vascular alterations are parallelled by clinical observations demonstrating similar features. Treatment strategies of reducing no-reflow after acute myocardial infarction are under investigation. Coronary microembolisation is significantly involved in clinically observed microvascular dysfunction, and new interventional devices provide hope for capturing these emboli before they cause tissue damage. From a practical standpoint, the best way to reduce no-reflow is to reduce infarct size by early reperfusion and adequate pharmacological treatment. However, whether improvement of microvascular perfusion even in zones of irreversibly damaged myocardium—which theoretically might have beneficial effects on ventricular remodelling, infarct healing, and collateral formation—is feasible with pharmacological interventions, remains to be investigated.
REFERENCES
Supplementary materials
Web only references
Angiographic assessment of no-reflow
-
Gibson CM, Murphy SA, Rizzo MJ, et al. Relationship between TIMI frame count and clinical outcomes after thrombolytic administration. Thrombolysis in Myocardial Infarction (TIMI) Study Group. Circulation 1999;99:1945-1950.
-
van�t Hof AW, Liem A, Suryapranata H, et al. Angiographic assessment of myocardial reperfusion in patients treated with primary angioplasty for acute myocardial infarction: myocardial blush grade. Zwolle Myocardial Infarction Study Group. Circulation 1998;97:2302-2306.
Myocardial contrast echocardiography
-
Ito H, Iwakura K. Assessing the relation between coronary reflow and myocardial reflow. Am J Cardiol 1998;81:8G-12G.
-
Lepper W, Hoffmann R, Kamp O, et al. Assessment of myocardial reperfusion by intravenous myocardial contrast echocardiography and coronary flow reserve after primary percutaneous transluminal angioplasty in patients with acute myocardial infarction. Circulation 2000;101:2368-2374.
-
Ito H, Iwakura K, Oh H, et al. Temporal changes in myocardial perfusion patterns in patients with reperfused anterior wall myocardial infarction. Their relation to myocardial viability. Circulation 1995;91:656-662.
-
Sakuma T, Otsuka M, Okimoto T, et al. Optimal time for predicting myocardial viability after successful primary angioplasty in acute myocardial infarction: A study using myocardial contrast echocardiography. Am J Cardiol 2001;87:687-692.
-
Karila-Cohen D, Czitrom D, Brochet E, et al. Decreased no-reflow in patients with anterior myocardial infarction and pre-infarction angina. Eur Heart J 1999;20:1724-1730.
Magnetic resonance imaging
-
Judd RM, Lugo-Olivieri CH, Arai M, et al. Physiological basis of myocardial contrast enhancement in fast magnetic resonance images of 2-day-old reperfused canine infarcts. Circulation 1995;92:1902-1910.
-
Rochitte CE, Kim RJ, Hillenbrand HB, et al. Microvascular integrity and the time course of myocardial sodium accumulation after acute infarction. Circ Res 2000;87:648-655.
Scintigraphic techniques
-
Jeremy RW, Links JM, Becker LC. Progressive failure of coronary flow during reperfusion of myocardial infarction: documentation of the no reflow phenomenon with positron emission tomography. J Am Coll Cardiol 1990;16:695-704.
-
Kondo M, Nakano A, Saito D, et al. Assessment of "microvascular no-reflow phenomenon" using technetium-99m macroaggregated albumin scintigraphy in patients with acute myocardial infarction. J Am Coll Cardiol 1998;32:898-903.
Potential mechanisms of no-reflow
-
Engler RL, Schmid-Schonbein GW, Pavelec RS. Leukocyte capillary plugging in myocardial ischemia and reperfusion in the dog. Am J Pathol 1983;111:98-111.
-
Litt MR, Jeremy RW, Weisman HF, et al. Neutrophil depletion limited to reperfusion reduces myocardial infarct size after 90 minutes of ischemia. Evidence for neutrophil-mediated reperfusion injury. Circulation 1989;80:1816-1827.
-
Kolodgie FD, Virmani R, Frab A. Limitation of no reflow injury by blood-free reperfusion with oxygenated perfluorochemical (Fluosol-DA 20%). J Am Coll Cardiol 1991;18:215-223.
-
Hale SL, Hammerman H, Kloner RA. Effect of two perfluorocarbon emulsions on reperfusion injury after coronary artery occlusion in rabbits. Basic Res Cardiol 1995;90:404-409
-
Li XS, Wang QD, Pernow J. Beneficial effects of the endothelin receptor antagonist bosentan on myocardial and endothelial injury following ischaemia/reperfusion in the rat. Eur J Pharmacol 1995;283:161-168.
-
Hale S, Kloner RA. Ischemic preconditioning in the rabbit: effect on regional myocardial blood flow. Coronary Artery Dis 1992;3:133-140.
-
Bauer B, Simkhovich BZ, Kloner RA, et al. Does preconditioning protect the coronary vasculature from subsequent ischemia/reperfusion injury? Circulation 1993;88:659-672.
-
Kloner RA, Giacomelli F, Alker KJ, et al. Influx of neutrophils into the wall of large epicardial coronary arteries in response to ischemia/reperfusion. Circulation 1991;84:1758-1772.
-
Manciet LH, Poole DC, McDonagh PF, et al. Microvascular compression during myocardial ischemia: mechanistic basis for no-reflow phenomenon. Am J Physiol 1994;266:H1541-H550.
-
Carlson RE, Aisen AM, Buda AJ. Effect of reduction in myocardial edema on myocardial blood flow and ventricular function after coronary reperfusion. Am J Physiol 1992;262:H641-H648
-
Willerson JT, Watson JT, Hutton I, et al. The influence of hypertonic mannitol on regional myocardial blood flow during acute and chronic myocardial ischemia in anesthetized and awake intact dogs. J Clin Invest 1975;55:892-902.
-
Siminiak T, Flores NA, Sheridan DJ. Neutrophil interactions with endothelium and platelets: possible role in the development of cardiovascular injury. Eur Heart J 1995;16:160-170.
-
Beauchamp P, Richard V, Tamion F, et al. Protective effects of preconditioning in cultured rat endothelial cells: Effects on neutrophil adhesion and expression of ICAM-1 after anoxia and reoxygenation. Circulation 1999;100:541-546.
-
Ohnishi M, Yamada K, Morooka S, et al. Inhibition of P-selectin attenuates neutrophil-mediated myocardial dysfunction in isolated rat heart. Eur J Pharmacol 1999;366:271-279.
-
Birnbaum Y, Patterson M, Kloner RA. The effect of CY1503, a sialyl Lewisx analog blocker of the selectin adhesion molecules, on infarct size and "no-reflow" in the rabbit model of acute myocardial infarction/reperfusion. J Mol Cell Cardiol 1997;29:2013-2025.
-
Golino P, Maroko PR, Carew TE. Efficacy of platelet depletion in counteracting the detrimental effect of acute hypercholesterolemia on infarct size and the no-reflow phenomenon in rabbits undergoing coronary artery occlusion-reperfusion. Circulation 1987;76:173-180.
-
Galiuto L, DeMaria AN, del Balzo U, et al.. Ischemia-reperfusion injury at the microvascular level treatment by endothelin A-selective antagonist and evaluation by myocardial contrast echocardiography. Circulation 2000;102:3111-3116.
-
Ambrosio G, Becker LC, Hutchins GM, et al. Reduction in experimental infarct size by recombinant human superoxide dismutase: insights into the pathophysiology of reperfusion injury. Circulation 1986;74:1424-1433.
-
Villari B, Ambrosio G, Golino P, et al. The effects of calcium channel antagonist treatment and oxygen radical scavenging on infarct size and the no-reflow phenomenon in reperfused hearts. Am Heart J 1993;125:11-23
-
Ambrosio G, Tritto I. Reperfusion injury: experimental evidence and clinical implications. Am Heart J 1999;138:S69-75.
-
Kloner RA. Does reperfusion injury exist in humans? J Am Coll Cardiol 1993;21:537-545.
-
Kloner RA, Alker KJ. The effect of streptokinase on intramyocardial hemorrhage, infarct size, and the no-reflow phenomenon during coronary reperfusion. Circulation 1984;70:513-521.
-
Kloner RA, Alker K, Campbell C, et al. Does tissue-type plasminogen activator have direct beneficial effects on the myocardium independent of its ability to lyse intracoronary thrombi? Circulation 1989;79:1125-1136.
Coronary microembolization
-
Yao SK, Ober JC, McNatt J, et al. ADP plays an important role in mediating platelet aggregation and cyclic flow variations in vivo in stenosed and endothelium-injured canine coronary arteries. Circ Res 1992;70:39-48.
-
Erbel R, Heusch G. Coronary microembolization. J Am Coll Cardiol 2000;36:22-24.
-
Mutin M, Canavy I, Blann A, et al. Direct evidence of endothelial injury in acute myocardial infarction and unstable angina by demonstration of circulating endothelial cells. Blood 1999;93:2951-2958.
-
Mak KH, Challapalli R, Eisenberg MJ, et al., for the EPIC Investigators. Effect of platelet glycoprotein IIb/IIIa receptor inhibition on distal embolization during percutaneous revascularization of aortocoronary saphenous vein grafts. Am J Cardiol 1997;80:985-988.
-
Carlino M, De Gregorio J, Di Mario C, et al. Prevention of distal embolization during saphenous vein graft lesion angioplasty. Experience with a new temporary occlusion and aspiration system. Circulation 1999;99:3221-3223.
-
Koch KC, vom Dahl J, Kleinhans E, et al. Influence of platelet GPIIb/IIIa receptor antagonist on myocardial hypoperfusion during rotational atherectomy as assessed by myocardial Tc-99 sestamibi scintigraphy. J Am Coll Cardiol 1999;33:998-1004.
-
Topol EJ, Leya F, Pinkerton CA, et al., for the CAVEAT Study Group. A comparison of directional atherectomy with coronary angioplasty in patients with coronary artery disease. N Engl J Med 1993;329:221-227.
-
Holmes DR, Topol EJ, Califf RM, et al., for the CAVEAT-II Investigators. A multicenter, randomized trial of coronary angioplasty versus directional atherectomy for patients with saphenous vein bypass graft lesions. Circulation 1995;91:1966-1974.
Treatment strategies
-
Pitarys CJ, Virmani R, Vildibill HD, et al. Reduction of myocardial reperfusion injury by intravenous adenosine administered during the early reperfusion period. Circulation 1991;83:237-247.
-
Homeister JW, Hoff PT, Fletcher DD, et al. Combined adenosine and lidocaine administration limits myocardial reperfusion injury. Circulation 1990;82:595-608.
-
Marzilli M, Orsini E, Marraccini P, et al. Beneficial effects of intracoronary adenosine as an adjunct to primary angioplasty in acute myocardial infarction. Circulation 2000;101:2154-2159.
-
Flaherty JT, Pitt B, Gruber JW, et al. Recombinant human superoxide dismutase (h-SOD) fails to improve recovery of ventricular function in patients undergoing coronary angioplasty for acute myocardial infarction. Circulation 1994;89:1982-1991.
-
Ishihara M, Sato H, Tateishi H, et al. Attenuation of the no-reflow phenomenon after coronary angioplasty for acute myocardial infarction with intracoronary papaverine. Am Heart J 1996;132:959-963.
-
Hillegass WB, Dean NA, Liao L, et al. Treatment of no-reflow and impaired flow with the nitric oxide donor nitroprusside following percutaneous interventions: initial human clinical experience. J Am Coll Cardiol 2001; 37:1335-1343.
-
Neumann FJ, Blasini R, Schmitt C, et al. Effect of glycoprotein IIb/IIIa receptor blockade on recovery of coronary flow and left ventricular function after the placement of coronary-artery stents in acute myocardial infarction. Circulation 1998;98:2695-2701.
-
Gibson CM, Cohen DJ, Cohen EA, et al., for the ESPRIT study group. Effect of Eptifibatide on coronary flow reserve following coronary stent implantation (an ESPPRIT substudy). Enhanced Suppression of the Platelet IIb/IIIa Receptor with Integrilin Therapy. Am J Cardiol 2001;87:1293-1295.
-
Linked Articles
- Miscellanea
- Miscellanea