Article Text

Statistics from Altmetric.com
Rheumatoid factors (RFs) are autoantibodies against IgG. They are probably the most studied antibody since their discovery by Waaler in 1937. In this editorial we aim to briefly examine the proposed roles of RFs, how they may contribute to disease pathogenicity, and, in so doing, mention the application of novel peptide technology, crystallography and molecular modelling to rheumatoid factor study.
Occurrence and roles of rheumatoid factors
RFs are not exclusive to rheumatoid arthritis (RA). They are found in a number of other autoimmune diseases, such as systemic lupus erythematous and Sjögren’s syndrome, infectious diseases such a mycobacterium tuberculosis, and Lyme disease and in healthy people.1
RFs seem to have a beneficial role to the normal functioning of the immune system. They may do this in a number of ways:
(1) Clearance of immune complexes. IgM and IgG RFs may bind to an antibody-antigen complex and facilitate clearance by binding to the Fc receptors on phagocytes.2 In particular, this may facilitate clearance of IgG2 and IgG4 immune complexes as these subclasses do not readily activate complement.
(2) Antigen processing by B cells. B cells can act as antigen presenting cells as they can use their surface immunoglobulin to capture antigen. Membrane bound RFs can therefore capture immune complexes containing IgG that can then be internalised, which may lead to the presentation of a relevant epitope to a T cell as part of an immune response.2 ,3
(3) Setting up an early antibody repertoire. Antibodies that behave as RFs can be found in human fetal liver and spleen,4 ,5 which suggests that autoreactive B cells are playing a part in the development of a functional antibody repertoire.
How can rheumatoid factors contribute to disease pathogenesis?
RF involvement in immune complex formation may lead to fixation of complement and recruitment of macrophages, neutrophils, and lymphocytes. Such involvement may be one part of an autoimmune inflammatory response that results in arthritis.
So how can RFs on the one hand be beneficial while on the other be potentially pathogenic? One clue we have is that naturally occurring RFs are mainly of the IgM isotype, while IgG RFs are thought to be associated with disease. It is probable that the production of pathogenic RFs are dependent on a class switch of antibody from IgM to IgG. This would imply that T cell help is available as the switch from IgM to IgG is usually under T helper cell control. This leads us to question ‘what are the antigenic peptides that activate T cells and allow them to help B cells produce IgG RFs?’. Identifying such a peptide trigger has been elusive. It may be that one has to look at peptides from IgG itself or even RFs. In support of this notion, there are data to suggest that κIIIb light chains from RFs can cause T cell proliferation (J Petersen et al, 7th international conference on immunology, 1989).
The mechanism of rheumatoid factor binding to IgG
There is at least 6 mg/ml of IgG in the serum and only 3 mg/ml of IgM. If all this were RF, which is unlikely, it is surprising that we can detect free RF at all in such an excess of IgG antigen. This suggests that either some RFs do not bind to certain types of IgG or that not all IgG is of a type that is capable of being bound by RFs.
The site of binding for many RFs is thought to lie at the Cγ2-Cγ3 domain interface in Fc, based upon evidence from serology, human subclass reactivity patterns, and more recently, site directed mutagenesis.6 The last approach has identified key determinants in discontinuous loop regions of Cγ2 and Cγ3. Yet it is not known whether pathogenic and non-pathogenic RFs bind to similar or separate sites on the IgG molecules.
It is known that in RA IgG oligosaccharides change such that there is a shift to the agalactosylated form,7 which may uncover novel peptide epitopes and hence facilitate RF binding. Using monoclonal RFs from synovial tissue, it has recently been demonstrated that in some instances the binding is greatly increased when galactose is absent.8 These same RFs had at least 10 times higher affinity for human IgG9 than those monoclonal RFs that bound independently of sugar.
The work of Hay and others have suggested that there may be several mechanisms for RF binding.10
(1) Binding to continuous or discontinuous determinants on IgG away from the influence of oligosaccharides.11
(2) Binding to peptides that have been uncovered by the absence of oligosaccharide or made available for interaction because of conformational change resulting from oligosaccharide absence.12
(3) Binding to IgG via RF framework peptides away from the conventional complementarity determining regions.13-15
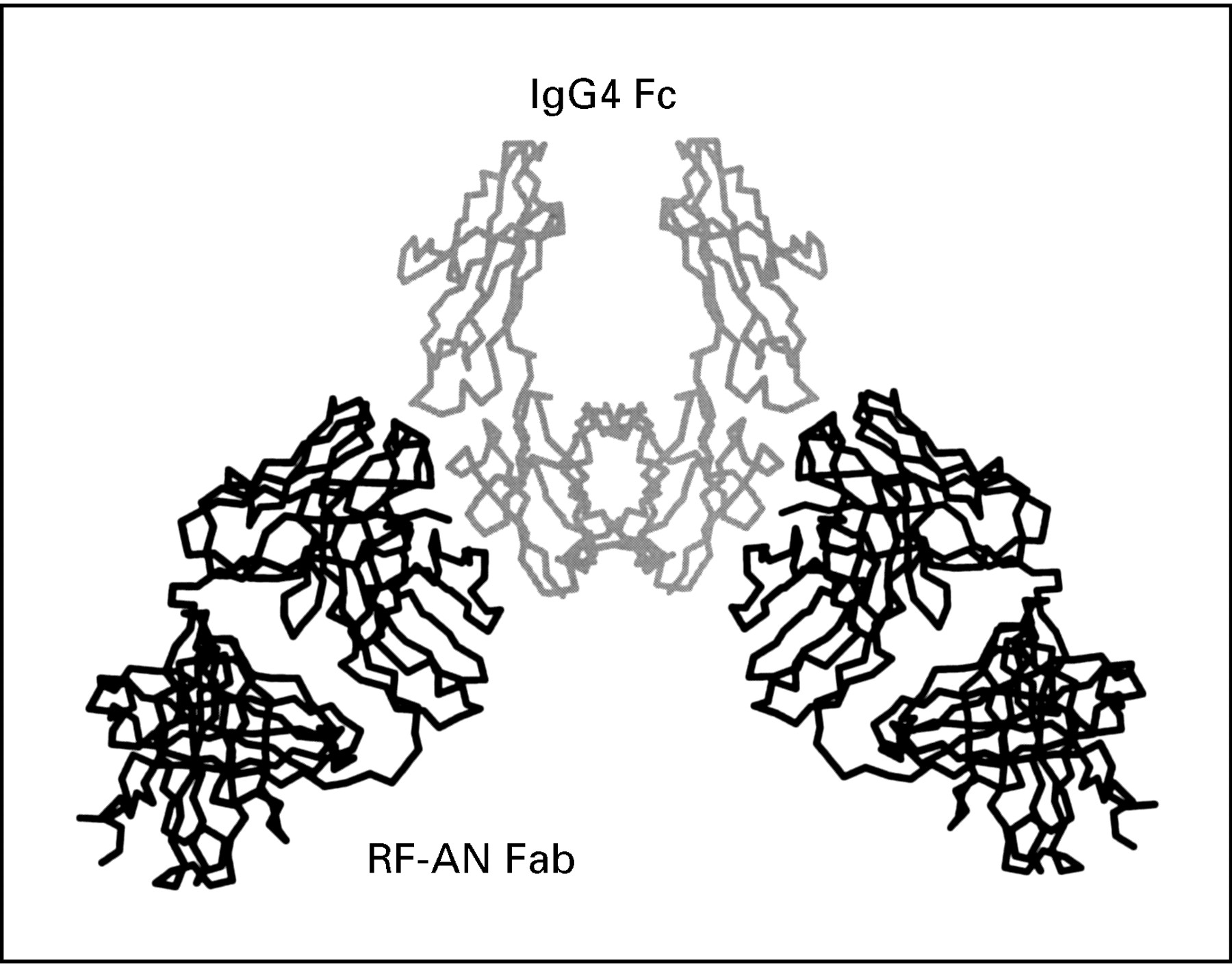
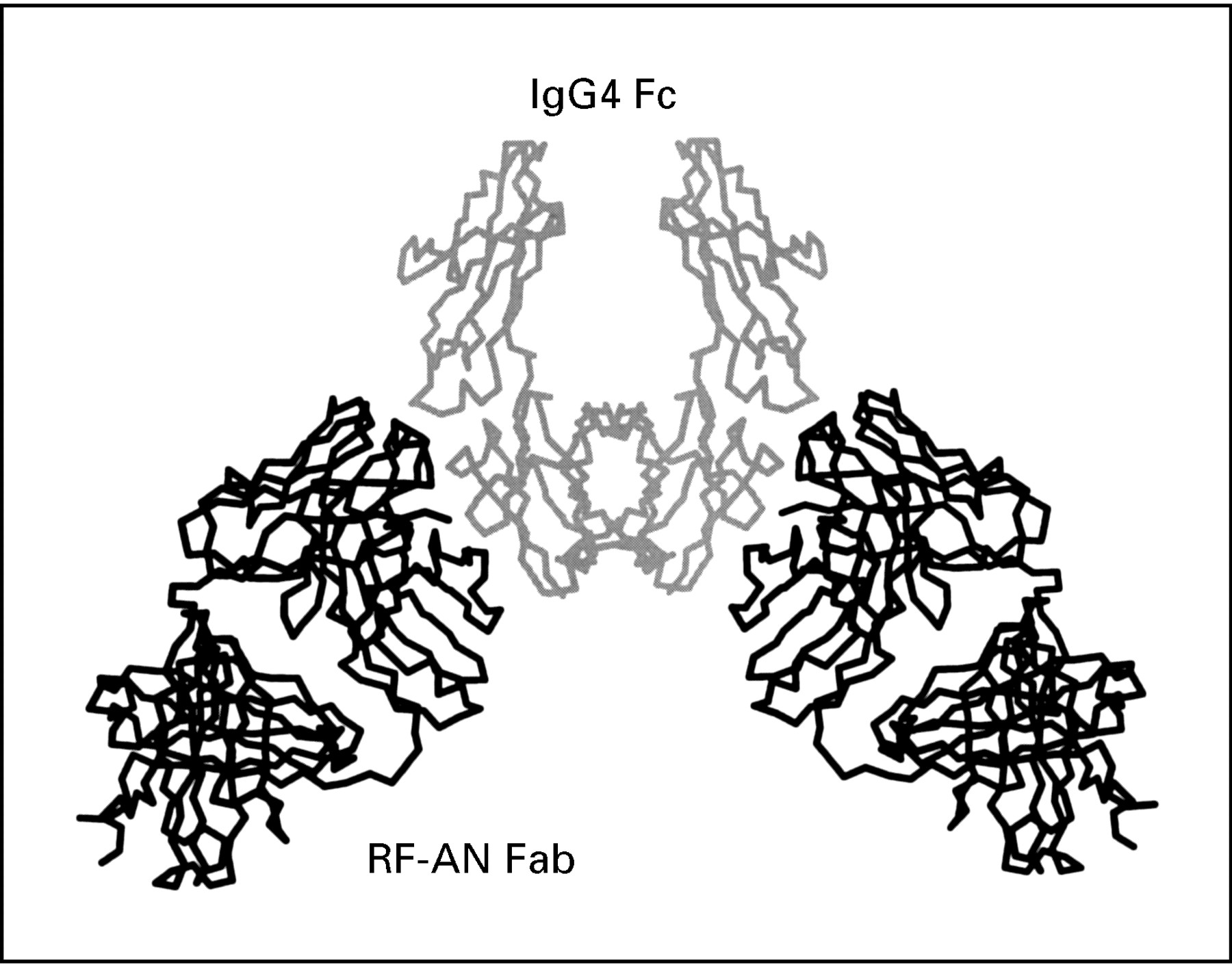
The structure of the first complex between a RF and IgG Fc has now been determined by x ray crystallography16 17 and it shows the RF Fab fragment binding through its complementarity determining regions to an epitope on the IgG Fc that spans the Cγ2 and Cγ3 domains (see fig 1). The RF in this study (RF-AN) is derived from the peripheral B cell of an RA patient18 and is an IgM/ λ antibody with a pattern of reactivity with human subclasses IgG 1, 2, and 4 that is typical of many RFs and mirrors that of the bacterial Fc-binding protein A. The binding site for protein A lies in the cleft between the two domains,19 and this RF clearly overlaps this area. The human IgG4 Fc present in the complex is predominantly agalactosyl,16 ,20 similar to that found in the immune complexes of RA patients. However, for this particular RF, the carbohydrate itself does not seem to be involved in RF recognition of Fc.
{kind=link}
Diagramatic representation of rheumatoid factor AN Fab fragment binding to an epitope in IgG Fc that spans the Cγ2 and Cγ3 domains as determined by x ray crystallography.
It is now important to study the structures of the complexes formed by other higher affinity RFs, and of the IgG class, to compare the modes of interaction, the nature of the epitopes recognised, and the role of the oligosaccharides. It may then be possible to identify the structural features that distinguish the pathogenic from the non-pathogenic RFs.
Our further understanding of the mechanisms of RF binding may be diagnostically helpful if pathogenic RFs can be differentiated between those that are harmless. It would also open up a number of further options whereby the RF binding mechanisms could be interrupted.
Conclusion
It is clear that RFs are more than markers of RA and dissecting out their physiological role and how they may be involved in pathological mechanisms will be exciting. Crystallographic analysis and molecular modelling of both polypeptide and oligosaccharide structures will undoubtedly aid this process, and epitope mapping has also been shown to be a powerful tool. Whether new diagnostics and novel therapeutic interventions will result remains to be seen, let us hope so, but hopefully we will not have to wait another 50 years for the answer.