Article Text

Abstract
Background Hereditary recurrent fevers (HRFs) are rare inflammatory diseases sharing similar clinical symptoms and effectively treated with anti-inflammatory biological drugs. Accurate diagnosis of HRF relies heavily on genetic testing.
Objectives This study aimed to obtain an experts’ consensus on the clinical significance of gene variants in four well-known HRF genes: MEFV, TNFRSF1A, NLRP3 and MVK.
Methods We configured a MOLGENIS web platform to share and analyse pathogenicity classifications of the variants and to manage a consensus-based classification process. Four experts in HRF genetics submitted independent classifications of 858 variants. Classifications were driven to consensus by recruiting four more expert opinions and by targeting discordant classifications in five iterative rounds.
Results Consensus classification was reached for 804/858 variants (94%). None of the unsolved variants (6%) remained with opposite classifications (eg, pathogenic vs benign). New mutational hotspots were found in all genes. We noted a lower pathogenic variant load and a higher fraction of variants with unknown or unsolved clinical significance in the MEFV gene.
Conclusion Applying a consensus-driven process on the pathogenicity assessment of experts yielded rapid classification of almost all variants of four HRF genes. The high-throughput database will profoundly assist clinicians and geneticists in the diagnosis of HRFs. The configured MOLGENIS platform and consensus evolution protocol are usable for assembly of other variant pathogenicity databases. The MOLGENIS software is available for reuse at http://github.com/molgenis/molgenis; the specific HRF configuration is available at http://molgenis.org/said/. The HRF pathogenicity classifications will be published on the INFEVERS database at https://fmf.igh.cnrs.fr/ISSAID/infevers/.
- genetic diagnosis
- pathogenicity classification
- hereditary recurrent fever
- molgenis
- infevers
Statistics from Altmetric.com
Introduction
Autoinflammatory diseases (AIDs) are conditions caused by a dysregulated innate immune system. Hereditary recurrent fevers (HRFs) are rare diseases that represent the most known group among the monogenic AIDS. They present as sterile systemic inflammatory and febrile attacks.1–6 Their overlapping clinical features include fever, myalgia, arthralgia, fatigue, skin rash and common laboratory features include elevated serum inflammatory markers such as C reactive protein and serum amyloid A protein. Their presentation is markedly heterogeneous, ranging from isolated recurrent fever to severe disorders complicated by sensorineural hearing loss or neurological manifestations or secondary amyloidosis.7 In the last few years, it became evident that identification of the causative genetic abnormality at the earliest possible stage is crucial for proper HRF management, including rapid application of effective treatments and appropriate genetic counselling.
Genetic testing is widely implemented for HRF diagnosis.8–11 The availability of next-generation sequencing (NGS), allowing simultaneous investigation of multiple HRF-associated genes has greatly improved HRF genetic diagnosis.12–15 However, as the number of variants detected by this method is much higher when compared with previous methods such as Sanger sequencing, the genetic diagnosis has become more complex.
The interpretation of genetic results is often a challenge: when the available knowledge for a particular DNA variant is insufficient, or when variants in unexpected genes are identified. Another interpretation dilemma concerns minor allele frequency variants inevitably reported with several autoinflammatory phenotypes. In daily practice, consultation of ClinVar (the reference database for the clinical significance of human genetic variants), INFEVERS (the reference database for variants in AID-associated genes) and some in silico prediction tools (AGVGD, Sorts Intolerant From Tolerant (SIFT), Polyphen-2 and Combined Annotation Dependent Depletion (CADD) score)16–18 are usually performed. However, conflicting results and discrepancies have been found in ClinVar19 and following in silico predictions. In particular, referencing these sources for HRF-associated genes showed that a clear interpretation of pathogenicity was retrieved for a limited number of variants. Preliminary recommendations assigning clinical significance to frequently encountered DNA variants in HRFs have been published in 2012, but they too are limited to a small number of frequent variants.10 Table 1 shows representative examples of equivocal variant interpretation using these sources. Hence, none of the currently available resources is accurate and comprehensive enough to be adapted to the HRF patient management.
Representative examples of equivocal variant interpretation for HRF genes
Estimation of the clinical significance of genetic variants provided as a pathogenicity classification is a recent and expanding practice in the field of rare diseases. To this objective, many recommendations have been developed among which the American College of Medical Genetics and Genomics (ACMG) is the most commonly used.20 Next to general guidelines, the need for data sharing and a collaborative effort in the interpretation of variants in HRF-associated genes is highly warranted.
Here, we report on a consensus-driven pathogenicity classification of variants in HRF-associated genes carried out by an international panel of genetics experts in AIDs, based on published as well as unpublished experience. We developed a novel and innovative protocol adapted from the Delphi approach21 and built on a study showing benefit in data sharing for resolving discrepant interpretations of variants pathogenicity.22 This included repeated steps of expert review and classification rounds, finally resulting in a consensus for the large majority of variants. The consensus classification can be used as the current gold standard or reference for the four best characterised HRF-associated genes: MEFV, TNFRSF1A, NLRP3 and MVK, which are responsible for familial Mediterranean fever (FMF), tumour necrosis factor (TNF) receptor-associated periodic syndrome (TRAPS), cryopyrin-associated periodic syndromes (CAPS) and mevalonate kinase deficiency (MKD), respectively. The outcome of this collaboration will be easily accessible to geneticists, clinicians and scientific community on the INFEVERS database at: https://fmf.igh.cnrs.fr/ISSAID/infevers/.
Methods
The scoring expert panel
International geneticists recognised in the field of HRFs were invited to participate in the project. Eight centres from the Netherlands (MEVG), Italy (IC), Israel (YS), Spain (JIA), France (GS and IT), the UK (DR and EO), Turkey (BP), and the US (HMH) involved in HRF gene discovery and/or INFEVERS variant curation and/or quality assessment through the European Molecular Genetics Quality Network reviewed and classified the variants. All participants use NGS-based technologies in their routine clinical practice.
Classification methods and platform
The publicly accessible INFEVERS database includes genetic information on the variants reported or published on HRF genes. To develop a user-friendly system for the variant scoring project, we first extracted the minimal available genetic data (name of the gene, protein variant name, and cDNA change according to the Human Genome Variant Society nomenclature) for each of the four best known HRF-associated genes (MEFV: NM_000243.2; TNFRSF1A: NM_001065.3; NLRP3: NM_001243133.1; MVK: NM_000431.3).
These data were then downloaded into the MOLGENIS scientific data platform,23 which we configured for this project with methods to support variant classification, comparison and cooperative consensus building. Population frequencies of each variant were made available to help classification scoring. The experts could provide complete variant classification lists in a batch, or alternatively individually chose a pathogenicity classification value from a scrolling list after signing in. The classification fulfilled the ACMG recommendations20: (1) benign; (2) likely benign; (3) variant of uncertain significance (VOUS); (4) likely pathogenic; and (5) pathogenic. Variants were classified by combining evidence on their clinical pathogenicity from both (1) reports (publications and databases) and (2) the expert’s own unpublished studies on patients and families (functional studies, segregation analysis and in silico tools). In silico prediction tools consulted included AGVGD, SIFT, Polyphen-2 and CADD score.16–18 The scores given by the experts were then automatically compared and sorted by an algorithm specifically developed for the purpose, resulting in an updated overview on the consensus status of variant classification. MOLGENIS software is available for reuse at http://github.com/molgenis/molgenis, and the specific HRF configuration is available at http://molgenis.org/said/.
To facilitate the selection of variants for review during the iterative rounds, we introduced an intermediate step. Three groups were generated in MOLGENIS: consistent, compatible and opposite. Consistent: all experts came to the same or consistent (benign and likely benign, VOUS or pathogenic and likely pathogenic) classification. Compatible: there was a difference of at most one level (VOUS vs benign/likely benign, or VOUS vs pathogenic/likely pathogenic) across the experts. Opposite: there was a difference of more than one level in the three-class system (benign/likely benign vs pathogenic/likely pathogenic). Those groups were updated on real time at each round of scoring (see consensus process), and the compatible and opposite groups were selected for iterative rounds of reviewing (see figure 1).

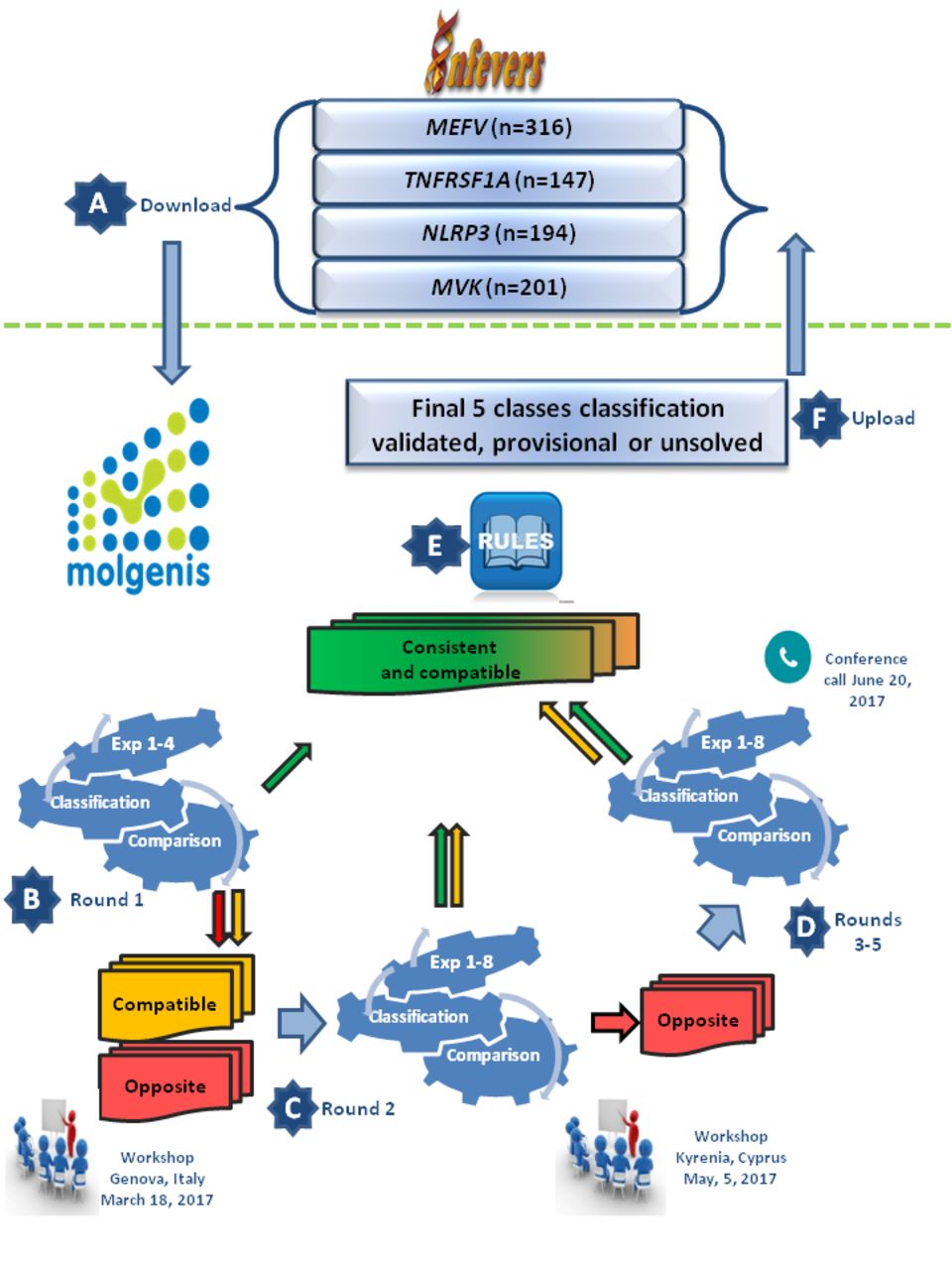{kind=link}
Flow representation of the different steps that were taken for HRF variant classification. (A) All HRF variants available as from March 2017 were downloaded from INFEVERS to the MOLGENIS platform. (B) Expert teams 1–4 blindly classified the variants according to the ACMG guidelines and their personal experience (round 1). (C) The four classifications were compared, and after a first workshop, all variants with ‘compatible’ or ‘opposite’ classification were reinvestigated by the four previous and four new experts (round 2). (D) After a second workshop, the remaining variants with ‘opposite’ classification were again revised (rounds 3–5). (E) A final classification of variants with ‘consistent’ or ‘compatible’ classifications was undertaken according to the majority vote and was tagged validated, or provisional, according to the consensus rule in table 2. Classification of some variants remained unsolved. (F) All variants classification results and their consensus tag will be reloaded into INFEVERS. ACMG, American College of Medical Genetics and Genomics; HRF, hereditary recurrent fever.
Consensus validation and classification rules
Each variant had to be classified by at least four independent experts. As requirement for unanimous voting would generate too many unsolved variants, a majority consensus was sought. We applied two rules—validation of consensus and five classes of majority classification—to define the consensus pathogenicity state of each variant. A classification was considered validated if ≥75% of the experts reached consistent votes. A provisional classification was assigned if between >50% and 75% of experts reached consistent votes. Variants that did not fulfil those criteria remained with the status ‘classification unsolved’. Table 2 shows that the actual majority rates for validated interpretations were higher than 75% when there were more than four evaluators. The second rule, applied consequently, assigned the majority vote as the final pathogenicity class to each solved variant. In case of equal number of votes, variants were classified into the lowest class (eg, likely benign if same number of benign and likely benign, or VOUS if any other compatible vote).
Validation rules
Interpretation workflow
Figure 1 summarises the steps we have taken to reach consensus classification. All the variants (n=858, INFEVERS, last update on March 2017) were downloaded to the MOLGENIS platform and evaluated blindly by four initial experts, namely MEVG, IT/GS, IC and JIA. The results obtained during this first round were discussed during a workshop of the European International Study Group for Systemic Autoinflammatory Diseases project held in Genoa, Italy, on 18 March 2017. As the percentage of variants without consensus was relatively high (>80%), and in order to increase the chances of achieving a consensual outcome for the variants with no straight forward interpretation or immediate consensus, it was decided to incorporate more geneticists into the classification panel. In a second blind scoring round, variants of the ‘compatible’ and ‘opposite’ groups were resubmitted via MOLGENIS to the four previous experts and to four new ones, namely DR/EO, BP, HMH and YS, leading to a revised content for the three groups. A second workshop was held during the International Society of Systemic Auto-Inflammatory Disease (ISSAID) conference in Kyrenia, Cyprus, on 5 May 2017, where it was decided to drive the opposite classifications to consensus. In the third classification round, the same procedure was followed, this time in visible mode for the ‘opposite’ group only, with the instruction to provide explanation and supporting evidence for each requalification. Once the experts were aware that their classification was out of consensus, they were free to proceed using their own way to re-evaluate the variants individually. This included a better read of the literature, more structured scoring, editing of mistakes and so on. The arguments provided by each expert, mainly based on unpublished data of familial segregation, functional data and recurrent association with disease, were shared and discussed during a wrap-up conference call on 20 June 2017. The fourth and fifth rounds were then carried out for the few remaining ‘opposite’ classifications, in the attempt to minimise their number. At the end of this process, each variant was scored by four to seven experts, the consensus rule and majority rule were applied, and individual classifications and their consensus rate (validated or provisional) were uploaded to the INFEVERS database.
Results
Consensus classification
Our workflow of iterative classification rounds left only 6% of the variants unsolved. Indeed, after applying the rules for consensus classification on each variant, we assigned a five-class classification to 803/858 (93.6%) variants, of which 372 had ‘consistent’ votes (46.3%) and 431 had ‘compatible’ votes (53.7%). A validated consensus classification was reached for 632 (74%) DNA variants. Provisional consensus classification was reached for 171 DNA variants (20%). Importantly, a validated consensus classification was reached for all pathogenic (n=119) and for 191 likely pathogenic variants, and none of the DNA variants remained with opposite classifications after all rounds.
Table 3 shows the classification results for each gene separately and reveals that 60%, 41% and 56% of the variants were validated as pathogenic or likely pathogenic in the MVK, NLRP3 and TNFRSF1A genes, respectively, compared with 9% in the MEFV gene. It is noteworthy that a high fraction of VOUS variants (96 out of 170; 56%) and most unsolved variants (37 out of 55; 67%) are MEFV variants, and none of the unsolved variants are TNFRSF1A variants.
See online supplementary file 1 for extensive lists of all the classified variants.
Supplementary file 1
The pathogenicity classifications of MEFV, MVK, NLRP3 and TNFRSF1A gene variants
Hotspots
New hotspots of likely pathogenic gene variations in adjacent amino acids of interest were drawn from the database (see online supplementary file 1). In the MEFV gene, four likely pathogenic variants translate to changes in amino acids p.Gly687 and p.Tyr688 within the PRYSPRY domain. In the TNFRSF1A gene, aside from multiple hotspots affecting cysteine and adjacent residues, two hotspots, one at p.Gly65, p.Thr66, p.Thr67 and p.Leu68 and the other at p.Asn94, p.His95 and p.Leu96, are suggested, each with five likely pathogenic variants. In the NLRP3 gene, 12 validated likely pathogenic variations span eight adjacent amino acids between p.Phe566 and p.Phe573, outside of the NACHT domain. The most common MVK pathogenic variant, p.Val377Ile, is surrounded by five likely pathogenic variants between p.Gly376 and p.His380.
Impact on clinical interpretation
The consensus-based pathogenicity scoring of variants in four HRF-associated genes described here allows us to propose a universal rule for genotype clinical interpretation of genetic test results according to the mode of disease inheritance (table 4).
Genotype interpretation according to the variant’s combination and mode of inheritance
The link between variant and genotype classification is direct for autosomal dominant conditions such as TRAPS and CAPS, for which only the presence of one pathogenic or likely pathogenic variant fully supports the clinical diagnosis (‘confirmatory genotype’). For recessive HRFs such as FMF and MKD, we suggest combining the pathogenicity score of the variants with zygosity. Only the presence of likely pathogenic or pathogenic variants on both alleles of the gene should result in a ‘confirmatory genotype’ and consequently a definitive diagnosis of the disease.
Discussion
Overall and gene-specific performance of consensus classification
We have created an online platform for a collaborative and comprehensive classification by multiple classifiers of DNA variants in the four best characterised HRF-associated genes for which a diagnosis is most frequently requested. We also generated a Delphi adapted protocol to reach a consensus among the experts. We sought a majority consensus rather than unanimous voting to avoid a high rate of unsolved variants. At the end of iterative classification rounds, a consensus classification based on at least consistent votes across experts was reached for 94% of the DNA variants. The sum result is capture and integration of current best knowledge into a gold standard classification reference.
Our scoring workflow aimed predominantly to reduce opposite classifications, known to occur even within the frame of applying the ACMG recommendations due to inconsistent implementation of various in silico prediction tools, allele frequency thresholds, functional data and medical publications.22 Indeed, publications can be conflicting as to whether certain published variants are true disease-causing mutations or benign polymorphisms. For this reason, some DNA variants are differently interpreted across the world, by laboratories and/or by clinicians.24 Such well-known examples in the HRF genes are c.442G>C encoding p.Glu148Gln in the MEFV gene,25 26 c.362G>A encoding p.Arg121Gln (historically named R92Q) in the TNFRSF1A gene27–29 and c.592G>A and c.2107C>A encoding p.Val198Met and p.Gln703Lys in the NLRP3 gene, respectively.30–32 Patel et al 33 recently configured a pathogenicity calculator that stores and summarises input collected from single or several classifiers, according to the ACMG guidelines. This transparency-based web tool, designed to organise and harmonise the methodology and resources of pathogenicity assessment, will help to sort conflicting interpretations, yet ultimately the final classification requires a process supporting and leading to expert consensus.
The opposite classifications of DNA variants among our HRF panel of experts that remained after the blinded classification rounds underwent discussions on the different lines of evidence during the visible steps of classification process. This resulted in no ‘opposite’ classifications at the end of the process.
With a consensus goal in mind, one would expect to see an increase in the number of variants classified as VOUS over the multiple rounds. However, only 20% of the variants ended with a VOUS classification (table 3). This is a significant achievement because the translation of pathogenic and benign classified variants back to the clinicians is easy, whereas the translation of VOUS classified variants is problematic. We believe that the high yield of classified variants reached in this work (94%) is attributable to in-depth knowledge of HRF-associated genes based on experience and familiarity with the literature and to prioritising gene disease-specific considerations. For instance, although there is no straightforward functional assay to assess the pathogenicity of rare TNFRSF1A variants, misfolding of TNFR is the generally accepted pathogenic mechanism that generates unfolded protein-mediated autoinflammatory response in TRAPS.34 Deleterious structural consequences on this protein are well predicted by in silico methods, and this probably explains the low fraction of VOUS and unsolved variants for the TNFRSF1A gene. For MVK gene variants, pathogenicity is ascertained by residual enzymatic activity or detection of elevated urinal mevalonic acid,5 6 and for NLRP3 variants by several inflammasome activation assays.32 35
An unforeseen outcome of this study is the many coding variants in MEFV voted with either VOUS or unsolved pathogenicity classification (42%, table 3), although the members of this consortium are all experienced in the key steps necessary for the interpretation of the HRF variants, that is, segregation analysis, matching genetic results to the phenotype of the patient after the test or functional assays. We believe that the underlying cause is current incomplete understanding of the molecular pathogenic mechanism responsible for FMF. Recent studies show that pyrin is a specific intracellular immune sensor for virulence activity, that is, bacterial modifications of Rho GTPases.36 The common pathogenic mutations causing FMF were shown to over-ride regulatory pathways affecting pyrin activity in knock-in mice.37 38 Yet insofar no functional assay correctly displays the known genotype–phenotype pathogenic correlation of MEFV variants in FMF. Perhaps consequently, in silico prediction tools such as Polyphen-2 or Align GVD do not accurately predict the pathogenicity of variants.39 For example, c.2080A>G encoding p.Met694Val in the MEFV gene is predicted to have a benign impact on protein function. Yet, this is the most frequent among all MEFV mutations and has the most severe clinical consequences. The population frequency of an MEFV variant is also less of a clue to evaluate its clinical significance. Recessive diseases such as FMF have spread in specific populations through a founder effect, drift and possible heterozygote advantage. In fact, the allele frequency of some variants is quite high. Conversely, rare variants are practically always encountered in solo (heterozygotes), and their clinical significance remains unknown (table 4).
Before ruling out clinical significance, it is important to take into account the possibility of dominant inheritance in FMF and/or other phenotypes associated with the MEFV gene. Most pathogenic or likely pathogenic mutations in the B30.2/SPRY domain (exon 10) are responsible for the classical form of FMF.1 2 36 40 Four mutations in exon 8, all affecting pyrin amino acid 577 putative coiled-coil domain, cause colchicine-responsive autosomal dominant AID resembling FMF.41 42 Finally variants in exon 2 of the MEFV gene, resulting in the loss of a 14-3-3 binding motif, were identified in patients with dominant pyrin-associated autoinflammation with neutrophilic dermatosis presenting with recurrent episodes of neutrophilic dermatosis, fever, elevated acute phase reactants, arthralgia and myalgia/myositis.43 44 This disease resembles pyogenic arthritis, pyoderma gangrenosum and acne syndrome, an AID caused by mutation in the PSTPIP1 gene.45
Confirming clinical diagnosis
The type, the number and the parental origin of the mutations must be taken into account for genetic confirmation of the diagnosis. While some genotypes are unambiguous and clearly pathogenic (eg, p.Met694Val homozygosity for FMF), the qualification of ‘confirmatory’ or ‘not confirmatory’ genotype remains sometimes variable according to the experience of the laboratory. We elaborated simple rules (table 4) that should provide an objective and reproducible classification for the majority of HRF genotypes. As discussed, particular attention should be paid to the selection of the correct disease inheritance mode for MEFV variants. Moreover, some MVK variants are classified as pathogenic in both a recessive mode such as MKD, and a dominant mode such as in disseminated superficial actinic porokeratosis diseases (eg, MVK: c.417dupC encoding p.Gly140Argfs*47, c.604G>A encoding Gly202Arg and c.1126G>A encoding p.Gly376Ser).46 Therefore, the disease that is clinically suspected should be considered before applying the genotype rules.
Conclusions
This work demonstrates that sharing of DNA variant classifications worldwide, and collaborative efforts to discuss lines of evidence, minimises misinterpretation of DNA variants in HRF genes. Periodic review and implementation of future relevant knowledge are intended to keep the current classifications updated and other hereditary AID’s variants will be submitted to the publicly available INFEVERS database.
The result of the genetic test was considered the most important criterion for disease diagnosis in a Delphi survey on the differential diagnosis and classification of the four HRFs (Gattorno et al, 2018, unpublished). A need for consensus evidence-based criteria on the basis of the combination of clinical and genetic features was proposed for the diagnosis of FMF.11 Hence, disease classification and diagnosis may strictly rely on the conclusion given in the report issued by the geneticist. We believe these gold standard classifications will dramatically speed up and synchronise HRF diagnosis between geneticists and clinicians in particularly in the context of increasing NGS data and will also direct efforts to close knowledge gaps. The MOLGENIS platform and consensus evolution protocol can support the assembly of other DNA variants pathogenicity databases.
Key messages
A consensus on the interpretation of hereditary recurrent fever (HRF) genetic variants is essential, because HRF diagnosis heavily relies on genetic testing.
An expert panel solved the pathogenicity of almost all genetic variants in four well-known HRF genes, after iterative classification rounds.
The clinical diagnosis of a monogenic HRF is genetically confirmed by presence of pathogenic and/or likely pathogenic variant(s) in the associated gene: one variant in the case of a dominant HRF and two bialellic variants in the case of a recessive HRF.
Acknowledgments
We would like to thank the support from the MOLGENIS software team and data team of the Genomics Coordination Center of the University Medical Center Groningen, in particular Joeri van der Velde, Bart Charbon, Mark de Haan, Dennis Hendriksen, Fleur Kelpin, Tommy de Boer, Connor Stroomberg and Marieke Bijlsma.
References
Footnotes
MEVG, IC and YS contributed equally.
Contributors Design of the study: MEVG, IT, MAS and IC; design of databases: ECC, MS, MAS and FM. Data analysis: IT, MEVG, MS and ECC. Classifying variants: MEVG, IC, YS, JIA, GS, DR, EO, BP, HMH and IT. Writing the manuscript: IT, MEVG, IC and YS; revising the manuscript: JIA, DR, EO, MAS and HMH; all authors approved the manuscript.
Funding This study is part of the INSAID project funded by an E-Rare-3 program, grant number 9003037603. BBMRI-NL is a research infrastructure financed by the Netherlands Organization for Scientific Research (NWO), grant number 184.033.111. CORBEL is an ESFRI cluster project in the European Union’s Horizon 2020 research and innovation programme under grant agreement no. 654248. RD-Connect has received funding from the European Union Seventh Framework Programme (FP7/2007–2013) under grant agreement no. 305,444 (RD-Connect).
Competing interests None declared.
Patient consent Detail has been removed from this case description/these case descriptions to ensure anonymity. The editors and reviewers have seen the detailed information available and are satisfied that the information backs up the case the authors are making.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement The datasets generated and/or analysed during the current study are available in the MOLGENIS repository at http://molgenis.org/said/ and the INFEVERS repository at https://fmf.igh.cnrs.fr/ISSAID/infevers/. MOLGENIS software is available for reuse at http://github.com/molgenis/molgenis