Article Text

Abstract
Objective This study aimed to report end-of-study results on efficacy and safety of secukinumab 150 mg through 5 years in patients with ankylosing spondylitis (AS; MEASURE 1 extension trial (NCT01863732)).
Methods After the 2-year core trial, 274 patients receiving subcutaneous secukinumab 150 or 75 mg (following intravenous loading or initial placebo treatment to 16/24 weeks) every 4 weeks were invited to enter the 3-year extension study. Dose escalation from 75 to 150 mg (approved dose) was allowed at or after week 156 based on the judgement of the treating physician. Assessments at week 260 (5 years) included Assessment of SpondyloArthritis international Society (ASAS) 20/40 and other efficacy outcomes. Data are presented as observed. Safety assessment included all patients who received ≥1 dose of study treatment.
Results Of the 274 patients who entered the extension study, 84% (230/274) completed 5 years of treatment. ASAS20/40 responses were 78.6/65.2%, Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) 50 response was 63.4% and mean (±SD) BASDAI total score was 2.6±1.76 with secukinumab 150 mg at 5 years. Improvements in efficacy outcomes were sustained through 5 years. A total of 82 patients on secukinumab 75 mg (56.2%) had their dose escalated to 150 mg after week 168; ASAS40, ASAS-PR, ASAS 5/6 and BASDAI50 responses were improved in patients whose dose was escalated from secukinumab 75 to 150 mg. Secukinumab was well tolerated with a safety profile consistent over the course of the study.
Conclusions Secukinumab 150 mg provided sustained efficacy across multiple domains of AS with a favourable and consistent safety profile through 5-year treatment. Over 50% of patients required dose escalation from 75 to 150 mg and efficacy improved in these patients.
- DMARDs (biologic)
- inflammation
- anti-TNF
- ankylosing spondylitis
- spondyloarthritis
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Anti-tumour necrosis factor (TNF) agents are widely used in the treatment of ankylosing spondylitis (AS) and secukinumab is the only biologic (interleukin-17A inhibitor) other than anti-TNF agents currently approved for the treatment of AS. Secukinumab has demonstrated rapid and sustained benefits in clinical and radiographic outcomes in AS with a favourable safety profile through 4 years of therapy.
What does this study add?
End-of-study results from the MEASURE 1 extension trial represent the longest available efficacy and safety data reported for secukinumab in AS, and are notable for demonstrating no evidence of decreasing efficacy and a favourable and consistent safety profile up to 5 years.
How might this impact on clinical practice?
The results demonstrate that secukinumab is a long-term treatment option for patients with AS for both biologic-naïve subjects and for those patients who experience an inadequate response or intolerance to anti-TNF agents.
Introduction
Ankylosing spondylitis (AS) is a chronic inflammatory disease characterised by progressive, irreversible, structural damage of the sacroiliac and spinal joints, formation of syndesmophyte and ankylosis of the vertebral column, disability, reduced physical function and quality of life (QoL).1–4 The prevalence of AS is reported to range from 0.1% and 1.4% of the population,5–7 and it is associated with significant morbidity and disability, and thus constitutes a major socioeconomic burden.8
Non-steroidal anti-inflammatory drugs are the first-line treatment of AS. However, treatment is hindered by the lack of efficacy of effectively all standard disease modifying antirheumatic drugs (DMARDs), including sulfasalazine or methotrexate.9 Tumour necrosis factor (TNF) blocking agents were successfully added to the treatment armament of AS and subsequently demonstrated prolonged efficacy.10 11 Nevertheless, discontinuation of TNF blockers has been shown to be associated with rapid relapse.12 Also, a major challenge with TNF inhibitor therapy has been loss of efficacy over time,11 with up to 40% of patients reported to not respond or tolerate the therapy.13
In chronic conditions such as AS, clinical evaluation of efficacy and safety of long-term treatment is important for informed treatment decision-making.14 Long-term health-related QoL and physical function through the control of disease activity and inflammation, as well as the prevention of progressive structural damage, are the key objectives of treatment in patients with AS.14 15
Secukinumab, a fully human monoclonal antibody that directly inhibits interleukin (IL)-17A, has shown significant and sustained efficacy in the treatment of AS across phase III MEASURE studies16–20 with a low rate of structural radiographic progression through 4 years in the MEASURE 1 study.16 21 Secukinumab 150 mg is the only approved biological DMARD for AS other than TNF inhibitors.2 Here, we present long-term (5 years), end-of-study efficacy and safety results for secukinumab from the MEASURE 1 trial in patients with AS, including results for patients whose dose was escalated from secukinumab 75 to 150 mg during the study.
Methods
Study design and patients
MEASURE 1 is a phase III, double-blind, randomised, placebo-controlled 2-year study, with 3-year extension. Study design and primary results have been previously reported.22 Patients were initially randomised to receive either intravenous secukinumab 10 mg/kg at baseline, week 2 and week 4, followed by subcutaneous secukinumab 150 mg (intravenous → 150 mg) or 75 mg (intravenous → 75 mg) every 4 weeks thereafter. Matched placebo was given on the same intravenous to subcutaneous dosing schedule. Placebo patients were re-randomised to secukinumab 150 mg subcutaneous or secukinumab 75 mg subcutaneous (placebo switchers) by week 16 (non-responders) or week 24 (responders). After the 2-year core trial (NCT01358175), patients receiving secukinumab 150 or 75 mg subcutaneous were invited to enter a 3-year extension trial (NCT1863732). Following a protocol amendment, dose escalation from secukinumab 75 to 150 mg was allowed at or after week 156 or later visits based on the clinical judgement of the treating physicians; patients were not allowed to switch to a lower dose once dose escalation occurred. Patients were unblinded at week 156 (or after approval and implementation of Protocol Amendment 1, whichever occurred first) and continued to receive the same secukinumab treatment in an open-label fashion. Patients were stratified according to previous anti-TNF therapy (patients who were naïve to anti-TNF therapy (anti-TNF–naïve) or those with a history of inadequate response or intolerance to one of these agents (anti-TNF–IR)).
Endpoints and assessments
Efficacy assessments at week 260 (5 years) included Assessment of SpondyloArthritis international Society (ASAS) criteria 20 response; ASAS40 response; ASAS5/6 response; Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) score; BASDAI50 response; Bath Ankylosing Spondylitis Functional Index (BASFI) score; Bath Ankylosing Spondylitis Metrology Index (BASMI) score; Short Form-36 Physical Component Summary (SF-36 PCS) score; ASAS partial remission (ASAS-PR) response; Ankylosing Spondylitis Disease Activity Score-C-reactive Protein (ASDAS-CRP) inactive disease response; the Functional Assessment of Chronic Illness Therapy fatigue questionnaire (FACIT-Fatigue) score; overall safety and tolerability. Assessments in patients whose dose was escalated from 75 to 150 mg included ASAS20, ASAS40, ASAS-PR, ASAS5/6 and BASDAI50 (up to 72 weeks after dose escalation).
Statistical analysis
Extension full analysis set (FAS) comprised of all patients enrolled in the extension study. Efficacy results are reported as observed data and included all patients who had available data at baseline and at each corresponding treatment visit. ASAS20/40 responses are reported for patients originally randomised to secukinumab 150 mg to show the full 5-year efficacy, and separately for all patients who entered the extension study in the secukinumab 150 mg group (ie, including patients originally randomised to secukinumab 150 mg and patients originally randomised to placebo who switched to secukinumab 150 mg at weeks 16 or 24 (placebo switchers)). For patients who discontinued during the period from week 212 to week 260 (ie, current treatment period for this analysis), the end of treatment visit (ie, final assessment 4 weeks after last study treatment) was considered as week 260. The dose escalation subset comprised of all patients from the extension FAS who received at least one dose of the escalated dose. Safety analyses included all patients who received ≥1 dose of secukinumab, and data are presented as exposure adjusted incidence rates (EAIRs) per 100 patient-years over the entire treatment period. Adverse events (AEs) were coded using the Medical Dictionary for Regulatory Activities V.21.0 preferred terms (PTs) (https://www.meddra.org/). Safety results are reported for Any secukinumab, which includes all patients who received at least one dose of secukinumab 75 or 150 mg in core or extension study. A patient with multiple occurrences of an AE was counted only once in the AE category.
Data were collected in accordance with Good Clinical Practice guidelines by the study investigators and were analysed by the sponsor. Data presented here, from the end of study analysis at week 260 (5 years), and were collected up to 16 March 2018 (last patient last visit).
Results
Patients
Of the 371 patients who enrolled in the core study, 62% (230/371) completed 5 years of treatment. Of the 274 patients who entered the extension study, 84% (230/274) completed 5 years of treatment. Of the 128 patients in the secukinumab 150 mg group (ie, including placebo switchers), 108 patients (84%) completed 5 years of treatment (figure 1). Demographic and baseline characteristics have been reported previously.16 21 Approximately 80% of patients who entered the extension study were anti-TNF–naïve and 83.5% (86/103) of these patients completed 5 years of treatment.

Patient disposition—up to 5 years (week 260). Secukinumab 75 mg arm includes patients whose dose was escalated to secukinumab 150 mg starting at week 168 (n=82). AE, adverse event; Pt, patient; N, number of randomised patients; n, number of patients with an event.
Efficacy
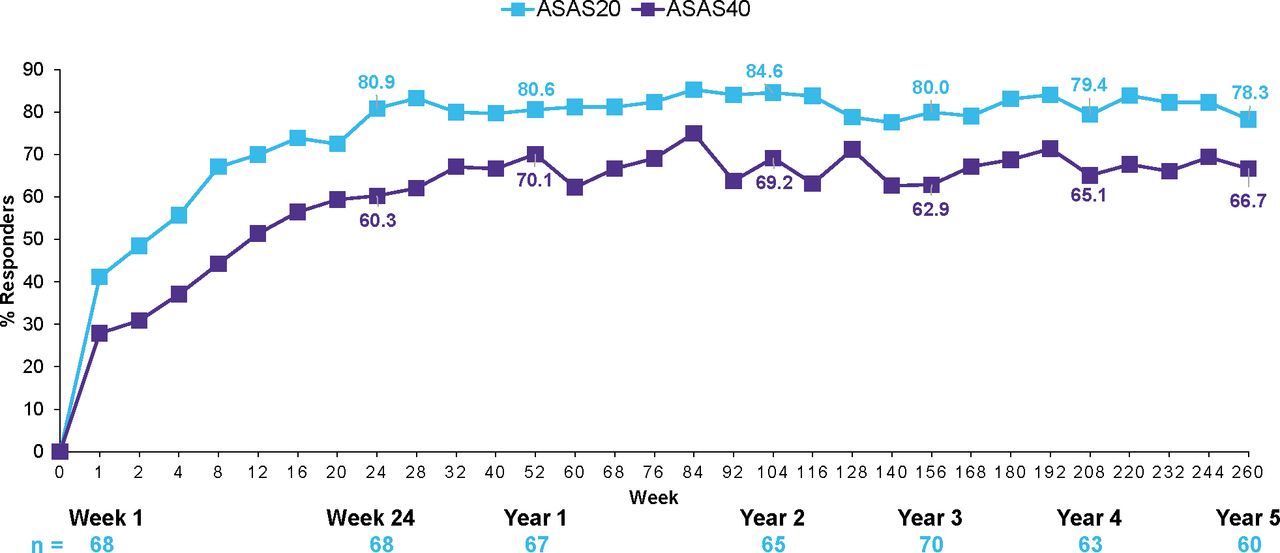
ASAS20 and ASAS40 responses were sustained through 5 years in patients who were originally randomised to secukinumab 150 mg and were 77.6% and 64.5%, respectively (figure 2). ASAS20 and ASAS40 responses with secukinumab 150 mg were also sustained through 5 years in the anti-TNF–naïve and anti-TNF–IR groups (figure 3).
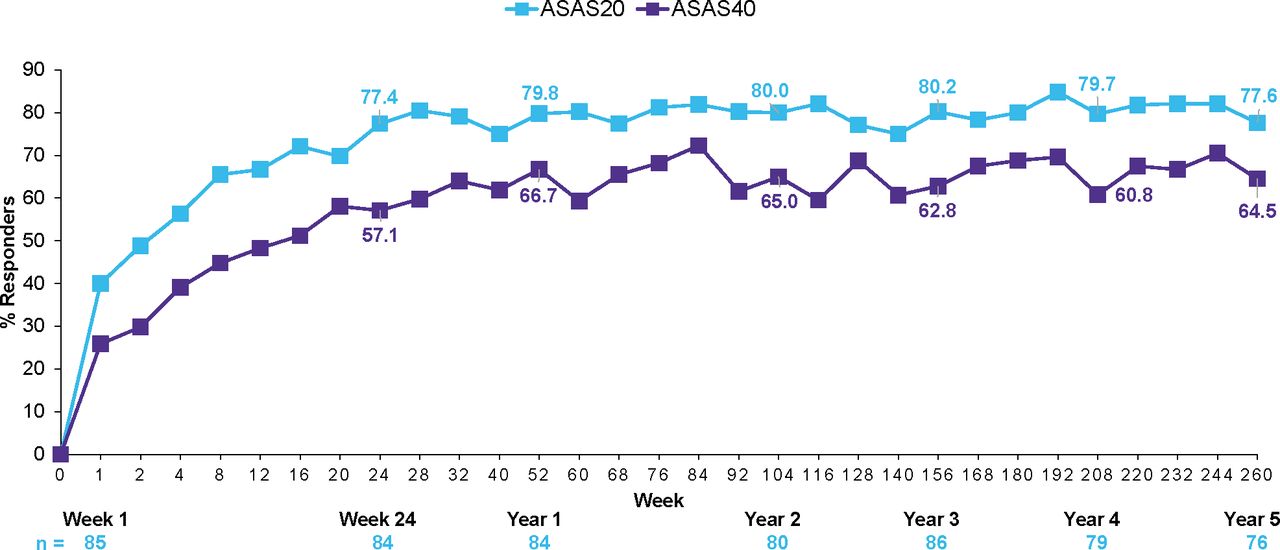
ASAS20/40 response rates through 5 years in secukinumab 150 group (n=87). Data shown are as observed through 5 years in patients originally randomised to secukinumab 150 mg without placebo switchers or patients whose dose was escalated. ASAS, Assessment of SpondyloArthritis international Society; N, number of patients randomised; n, number of patients assessed.

ASAS20/40 response rates in anti–TNF-naïve patients in secukinumab 150 group through 5 years (n=70). Data shown are as observed through 5 years in patients originally randomised to secukinumab 150 mg without placebo switchers or patients whose dose was escalated. ASAS, Assessment of SpondyloArthritis international Society; N, number of patients randomised, n, number of patients assessed; TNF, tumour necrosis factor.
In the overall group of patients that received secukinumab 150 mg (ie, including placebo switchers), ASAS20/40 responses at 5 years were 78.6/65.2%, with improvements sustained across all individual ASAS components (online supplementary table S1). Results for all other efficacy endpoints were also sustained through 5 years as shown in table 1.
Supplemental material
Summary of key efficacy outcomes through 5 years (week 260)
Efficacy following dose escalation
A total of 82 patients (56.2%) had their dose escalated from secukinumab 75 to 150 mg during the study, with the first patient having dose escalation beginning from week 168. ASAS20, ASAS40, ASAS-PR, ASAS 5/6 and BASDAI50 responses were improved in patients whose dose was escalated from secukinumab 75 to 150 mg (figure 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Efficacy responses in 82 patients whose dose was escalated from secukinumab 75 to 150 mg. Pre-escalation is defined as the last assessment done on or before the patient took the escalated dose. Postescalation results include patients who had reached each respective time period at the time of the analysis. ASAS, Assessment of SpondyloArthritis international Society; BASDAI, Bath Ankylosing Spondylitis Disease Activity Index; PR, partial remission.
Safety
The mean (±SD) exposure of Any secukinumab dose was 1446.1 (±631.2) days. The most common AEs (incidence rate >5 per 100 patient-years or relative frequency >2%) reported with Any secukinumab dose were nasopharyngitis, headache, diarrhoea and upper respiratory tract infection (URTI). The EAIRs of the most common AEs and selected AEs of interest are reported in table 2.
Summary of secukinumab safety at 5 years (week 260)
The infections were mainly nasopharyngitis and URTI. Candida infections (high-level term) were reported in a total of five patients in the secukinumab groups (three patients with oral candidiasis, one patient with Candida infection (PT) and one patient with genital candidiasis). Two opportunistic infections were reported in the Any secukinumab 75 mg group: herpes zoster cutaneous disseminated and tuberculosis (TB). The event of TB occurred ~2 months after the last dose of study treatment in a male patient with no medical history of TB and negative QuantiFERON TB test at screening. All the events of nasopharyngitis, URTI, Candida infections and opportunistic infections were of mild or moderate severity, non-serious and did not lead to study treatment discontinuation.
Over the 5-year treatment, nine patients were reported with inflammatory bowel disease (IBD) on Any secukinumab dose (seven patients with Crohn’s disease (PT; EAIR of 0.6 per 100 patient-years) and two patients with ulcerative colitis (PT; EAIR of 0.1 per 100 patient-years)), of which three patients had medical history of IBD and six patients reported de novo cases. For four patients (three patients with Crohn’s disease and one patient with ulcerative colitis), the IBD AE led to treatment discontinuation. Five of the seven cases of Crohn’s disease were reported during the 2-year core study; two further cases of Crohn’s disease and the two cases of ulcerative colitis were reported during the 3-year extension period.
Uveitis was reported in 24 patients (EAIR of 1.8 per 100 patient-years) across the two secukinumab treatment groups over the entire treatment period, of which 15 patients had a pre-existing history and 9 were de novo. One event in a patient receiving secukinumab 150 mg led to treatment discontinuation. Twelve of the 24 cases were reported during the core study and 12 during the 3-year extension period.
Treatment-emergent anti-drug antibodies (ADAs) were detected in one patient in the secukinumab 150 mg group during the extension study. ADAs were not associated with abnormal pharmacokinetic profiles or clinically relevant AEs.
Four deaths occurred during the 5-year treatment period (two during the core study: acute respiratory failure, suicide and two during the extension study: cerebrovascular accident, cardiac failure), neither of which was considered by the investigator to be related to study treatment.
Discussion
These results demonstrate that secukinumab 150 mg provided sustained efficacy and a favourable and consistent safety profile through 5 years of treatment in patients with AS, further building on previous evidence of sustained efficacy and low radiographic progression with secukinumab through 4 years.21
Clinical improvements were sustained across all endpoints through 5 years. More than half of the patients (82/146, 56.2%) originally randomised to secukinumab 75 mg required dose escalation to secukinumab 150 mg during the study based on the clinical judgement of the treating physicians, highlighting that the 75 mg dose was suboptimal for many patients.
These results represent the longest term results reported for an IL-17 inhibitor in AS and are notable for demonstrating sustained efficacy over 5 years. Over 80% of patients who entered the extension study completed 5 years of treatment, which is indicative of the sustained clinical benefit with long-term secukinumab therapy.
Clinical improvements with secukinumab were sustained in both anti-TNF–naïve and anti-TNF–IR subgroups. These results are consistent with the results of previous studies16–22 and provide additional supporting evidence for the use of secukinumab as a long-term treatment option for both biologic-naïve patients and those who experience an inadequate response or intolerance to anti-TNF agents.
In this cohort of secukinumab-treated patients with active AS, the exposure adjusted incidence rates of uveitis and IBD (new-onset cases and flares) were 1.8 and 0.5 per 100 patient-years, respectively, over 5 years of treatment with no increase over time. Secukinumab was well tolerated through 5 years with a safety profile consistent with previous reports through 5 years16–1821 22 and a recent pooled analysis of long-term data in patients with psoriasis, psoriatic arthritis and AS.23
Conclusions
Secukinumab provided sustained efficacy across multiple domains of AS, including signs and symptoms, physical function and objective markers of inflammation through 5 years. Efficacy improved in patients whose secukinumab dose was escalated from 75 to 150 mg (approved dose). The safety profile was consistent through 5 years of secukinumab treatment with no new or unexpected safety risks identified.
Acknowledgments
The authors thank the patients who participated in this study; the study investigators; and John Gallagher, a medical consultant working with Novartis. Medical writing and editorial support for this manuscript were provided by M K Vivek Sanker, Novartis, India, which was funded by Novartis Pharma in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3).
References
Footnotes
Contributors All named authors meet the International Committee of Medical Journal Editors criteria for authorship for this article, take responsibility for the integrity of the work as a whole, were involved in the drafting and critical review of the manuscript and approved the final version for submission. All authors agree to be accountable for all aspects of the work and attest to the accuracy and integrity of the work.
Funding The clinical study was sponsored by Novartis Pharma AG, Basel, Switzerland and designed by the scientific steering committee and Novartis personnel. Medical writing support was funded by Novartis.
Competing interests XB: Grant/research support from AbbVie, BMS, Celgene, Chugai, Merck, Novartis, Pfizer, UCB and Werfen. Consultant for AbbVie, BMS, Celgene, Chugai, Merck, Novartis, Pfizer, UCB and Werfen. Speakers bureau: AbbVie, BMS, Celgene, Chugai, Merck, Novartis, Pfizer, UCB and Werfen. JB: Grant/research support from AbbVie (Abbott), Amgen, BMS, Boehringer, Celgene, Celltrion, Centocor, Chugai, EBEWE Pharma, Medac, MSD (Schering-Plough), Mundipharma, Novartis, Pfizer (Wyeth), Roche, Sanofi-Aventis and UCB. Consultant for: AbbVie (Abbott), Amgen, BMS, Boehringer Ingelheim, Celgene, Celltrion, Centocor, Chugai, EBEWE Pharma, Medac, MSD (Schering-Plough), Mundipharma, Novartis, Pfizer (Wyeth), Roche, Sanofi-Aventis and UCB. Speakers bureau: AbbVie (Abbott), Amgen, BMS, Boehringer, Celgene, Celltrion, Centocor, Chugai, EBEWE Pharma, Medac, MSD (Schering-Plough), Mundipharma, Novartis, Pfizer (Wyeth), Roche, Sanofi-Aventis and UC. AD: Honoraria for consulting or speaking for, or has received research grants from AbbVie, Amgen, Boehringer Ingelheim, Bristol Myer Squibb (BMS), Eli Lilly, GlaxoSmithKline (GSK), Janssen, Novartis, Pfizer and UCB. DP: Grant/research support from AbbVie, MSD, Novartis and Pfizer. Consultant for AbbVie, BMS, Celgene, MSD, Novartis, Pfizer and UCB. Speakers bureau: AbbVie, BMS, Janssen, MSD, Novartis, Pfizer, Roche, UCB. AJK: Consultant for AbbVie, Pfizer, Genentech, UCB and Sanofi/Regeneron and Celgene. Speakers bureau: Celgene, Pfizer and Sanofi/Regeneron and Genentech. HT: Speakers bureau: Novartis, Eli Lilly and AbbVie. FVdB: Research grants, consultancy fees or speaker honoraria: AbbVie, BMS, Celgene, Galapagos, Janssen, Lilly, Merck, Novartis, Pfizer and UCB. EMD: Consultant for Novartis. ZT: Employee of Novartis with Novartis Stocks. AF: Employee of Novartis with Novartis Stocks.
Patient consent for publication Not required.
Ethics approval and consent to participate The clinical study was conducted in compliance with the Declaration of Helsinki, International Council for Harmonization Guidelines for Good Clinical Practice and local country regulations. All patients provided written informed consent to participate in the respective studies.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement The datasets generated and/or analysed during the current study are not publicly available. Novartis is committed to sharing with qualified external researchers’ access to patient-level data and supporting clinical documents from eligible studies. These requests are reviewed and approved based on scientific merit. All data provided are anonymised to respect the privacy of patients who have participated in the trial in line with applicable laws and regulations. The data may be requested from the corresponding author of the manuscript.