Article Text

Abstract
Introduction The current American College of Rheumatology and European League Against Rheumatism treatment recommendations advise tapering biological disease-modifying antirheumatic drug (bDMARD) therapy in patients with rheumatoid arthritis (RA) who achieve stable clinical remission while receiving bDMARDs. However, not all patients maintain remission or low disease activity after tapering or discontinuation of bDMARDs. The aim of the ImPact of Residual Inflammation Detected via Imaging TEchniques, Drug Levels and Patient Characteristics on the Outcome of Dose TaperIng of Adalimumab in Clinical Remission Rheumatoid ArThritis (RA) study, or PREDICTRA, is to generate data on patient and disease characteristics that may predict the clinical course of a fixed dose-tapering regimen with the bDMARD adalimumab.
Methods and analysis PREDICTRA is an ongoing, multicentre, phase IV, randomised, double-blind, parallel-group study of adalimumab dose tapering controlled by withdrawal in participants with RA who achieved stable clinical remission while receiving adalimumab. The study includes a screening period, a 4-week lead-in period with open-label adalimumab 40 mg every other week and a subsequent 36-week double-blind period during which participants are randomised 5:1 to adalimumab 40 mg every 3 weeks (taper arm) or placebo (withdrawal arm). The primary explanatory efficacy variables are lead-in baseline hand and wrist MRI-detected synovitis and bone marrow oedema scores, as well as a composite of both scores; the dependent variable is the occurrence of flare up to week 40. Additional efficacy variables, safety, pharmacokinetics, biomarkers and immunogenicity will also be assessed, and an ultrasound substudy will be conducted.
Ethics and dissemination The study is conducted in accordance with the International Conference on Harmonisation guidelines, local laws and the ethical principles of the Declaration of Helsinki. All participants are required to sign a written informed consent statement before the start of any study procedures.
Trial registration number EudraCT 2014-001114-26 and NCT02198651; Pre-results.
- rheumatology
- rheumatoid arthritis
- adalimumab
- withdrawal
- tapering
- discontinuation
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
The PREDICTRA study uses both MRI and ultrasound imaging techniques to allow a more comprehensive evaluation of musculoskeletal inflammation.
The randomised, double-blind, placebo-controlled study design reduces bias by eliminating expectations of treatment benefit or its loss based on treatment assignment.
Another strength of the study is that the open-label rescue arm provides an opportunity to assess the effectiveness of retreatment with the standard adalimumab regimen.
However, the duration of the trial may be insufficient to assess long-term progression of MRI-detected structural joint damage.
Other limitations include multiple testing bias when investigating several possible predictors and the reality that some measurements are costly or somewhat difficult to routinely do in an outpatient clinic.
Introduction
Rheumatoid arthritis (RA) affects approximately 1% of the population and imposes a significant economic burden, resulting from both direct and indirect costs.1–3 The primary goal of treating patients with RA under the treat-to-target recommendations is to maximise long-term health-related quality of life by controlling symptoms, preventing structural damage and normalising physical function and social participation.4 The most important way to achieve these goals is by abrogation of inflammation, with the treatment targets of clinical remission or low disease activity (LDA).4 Tumour necrosis factor inhibitor (TNFi) therapies effectively treat early and established RA, improving the disease course and halting structural progression.5 6 Adalimumab is a fully human immunoglobulin G1 antibody TNFi that was initially approved for the treatment of adult RA.7 It has since been approved for 13 additional indications.7 8
For patients with RA who achieve stable clinical remission with biological disease-modifying antirheumatic drug (bDMARD) therapy, the current American College of Rheumatology (ACR) and European League Against Rheumatism (EULAR) treatment recommendations advise cautiously tapering the bDMARD (ie, reducing the dose or increasing the interval spacing).9 10 However, not all patients maintain remission or LDA after tapering or discontinuation of biological therapy. Controlled withdrawal studies in patients with early RA have shown varying results for different TNFi agents. In the OPTIMA study in methotrexate-naive patients with early RA, 81% of patients who achieved stable LDA (Disease Activity Score based on 28 joints (DAS28) <3.2) at weeks 22 and 26 with adalimumab plus methotrexate maintained disease control for 52 weeks after discontinuing adalimumab.11 In contrast, in the PRIZE study in patients with early RA, only 69% of patients who withdrew etanercept but continued methotrexate therapy maintained DAS28 ≤3.2 after 39 weeks.12 In patients with established RA, withdrawing a TNFi or other bDMARD has been associated with RA flares in a higher proportion of patients than in those with early RA.13 Furthermore, some patients with RA failed to maintain LDA or clinical remission status when assessed at 48 weeks,14 52 weeks15 or up to 18 months16 following TNFi dose tapering.
Reliable parameters that would predict which patients can maintain adequate disease control after tapering or discontinuation of therapy could help guide treatment decisions. A number of studies over recent years have shown that ultrasound (US)-detected joint inflammation, in particular power Doppler–determined synovitis and to a lesser extent grayscale synovial hypertrophy (SH), are better predictors of relapse after bDMARD tapering or withdrawal than clinical measures.17–19 However, the added value of US to clinical examination at a patient level may still need to be confirmed.20 While MRI predictor data after bDMARD tapering or withdrawal are lacking, MRI-detected synovitis and bone marrow oedema have been demonstrated to predict radiographic progression in patients with RA,21 22 and MRI provides a reliable and responsive tool for multicentre studies (with central reading). In general, patients with early RA or those with deeper or longer clinical remission are less likely to experience disease flare after bDMARD tapering but further studies are needed to confirm these results.10 Thus, unanswered questions related to bDMARD dose tapering include: (1) what patient or disease characteristics are predictive of flare risk and (2) whether those characteristics could be used to devise a patient selection algorithm. Further, it is not well understood to what extent reinstitution of standard TNFi dosing after flare provides the same level of disease control as before the tapering.23 24 Although reinstitution of TNFi therapy has been successful for regaining disease control in the majority of patients (up to 100% of patients in some studies),18 23 25 not all studies have shown results that are as beneficial (regain of remission in only 57% of patients)26 and patient numbers were generally low. The imPact of Residual inflammation detected via imaging t Echniques, Drug levels and patient characteristics on the outcome of dose taperIng of adalimumab in Clinical remission rheumatoid arThritis (RA) study (PREDICTRA) was designed to address these questions, with a particular focus on the predictive value of MRI-detected inflammation.
Methods
Participants
Adult male or female participants ≥18 years of age with a diagnosis of RA (defined by the 1987 revised ACR classification criteria and/or the ACR/EULAR 2010 classification criteria) are eligible for study inclusion if they meet the following additional criteria: have been treated with adalimumab 40 mg every other week (eow) subcutaneously for ≥12 months before week 0 of the lead-in period and are in stable clinical remission defined by ≥1 documented Disease Activity Score based on 28 joints, erythrocyte sedimentation rate (DAS28(ESR))27 or DAS28 C reactive protein (CRP) of <2.6 (using four variables or three variables if patient global assessment (PGA) is unavailable) for ≥6 months before screening and DAS28(ESR) of <2.6 at screening (based on four variables).28 29 Concomitant methotrexate or other allowed conventional synthetic DMARDs (csDMARDs; eg, chloroquine, hydroxychloroquine, sulfasalazine, gold formulations and/or leflunomide) are permitted if the regimen has been stable for ≥12 weeks before week 0 of the lead-in period; the protocol allows for up to 20% of participants be included in the study if receiving stable doses of csDMARDs other than methotrexate or receiving adalimumab monotherapy, for at least 12 weeks before the week 0 lead-in visit. In addition, any concomitant oral corticosteroids (prednisone or equivalent) must have been administered at a dose of <10 mg/day that had been stable for ≥4 weeks before week 0 of the lead-in period and any nonsteroidal anti-inflammatory drugs must have been taken at a stable dose for ≥4 weeks before week 0 of the lead-in period. Exclusion criteria include any DAS28(ESR) or DAS28(CRP) of ≥2.6 that was assessed ≤6 months before screening, current use of bDMARDs other than adalimumab and any medical condition precluding contrast-enhanced MRI.
Study design and treatments
PREDICTRA is an ongoing, phase IV, multicentre, randomised, double-blind, parallel-group study of adalimumab dose tapering controlled with adalimumab withdrawal in patients with RA who achieved stable clinical remission while receiving adalimumab in the clinical setting (current protocol version from 25 February 2016; registered per the WHO Trial Registration Data Set at http://www.clinicaltrialsregister.eu (EudraCT 2014-001114-26) and clinicaltrials.gov (NCT02198651)). Study centres are located in Australia, Austria, Canada, France, Germany, Greece, Hungary, Ireland, Italy, the Netherlands, Spain, Sweden, UK and USA. The study includes a screening period of up to 28 days, a 4-week lead-in period with open-label adalimumab 40 mg administered subcutaneously eow and a subsequent 36-week double-blind period during which participants are randomised 5:1 to adalimumab 40 mg every 3 weeks (taper arm) or placebo (withdrawal arm; figure 1). Injections of study treatment are observed during clinic visits to ensure proper injection technique; participants record each administration in the clinic and elsewhere for compliance documentation. During the lead-in period, participants undergo a contrast-enhanced MRI scan of the most affected hand and wrist (or dominant hand and wrist if both sides are equally affected) before the double-blind baseline visit at week 4. At the double-blind baseline visit, only participants with DAS28(ESR) of <2.6 and an MRI scan quality-checked by a central reading centre are randomised; further study visits occur every 6 weeks until week 40. A flare during the double-blind period is defined as either DAS28(ESR) >2.6 and an increase of >0.6 in DAS28(ESR) from double-blind baseline or an increase of ≥1.2 from double-blind baseline DAS28(ESR) irrespective of absolute DAS28(ESR) score at any visit. If a flare occurs, that visit is denoted as the flare week 0 visit and the participant enters an open-label rescue arm with standard adalimumab dosing (40 mg eow) and is followed for 16 weeks (subsequent study visits on flare weeks 4, 10 and 16).

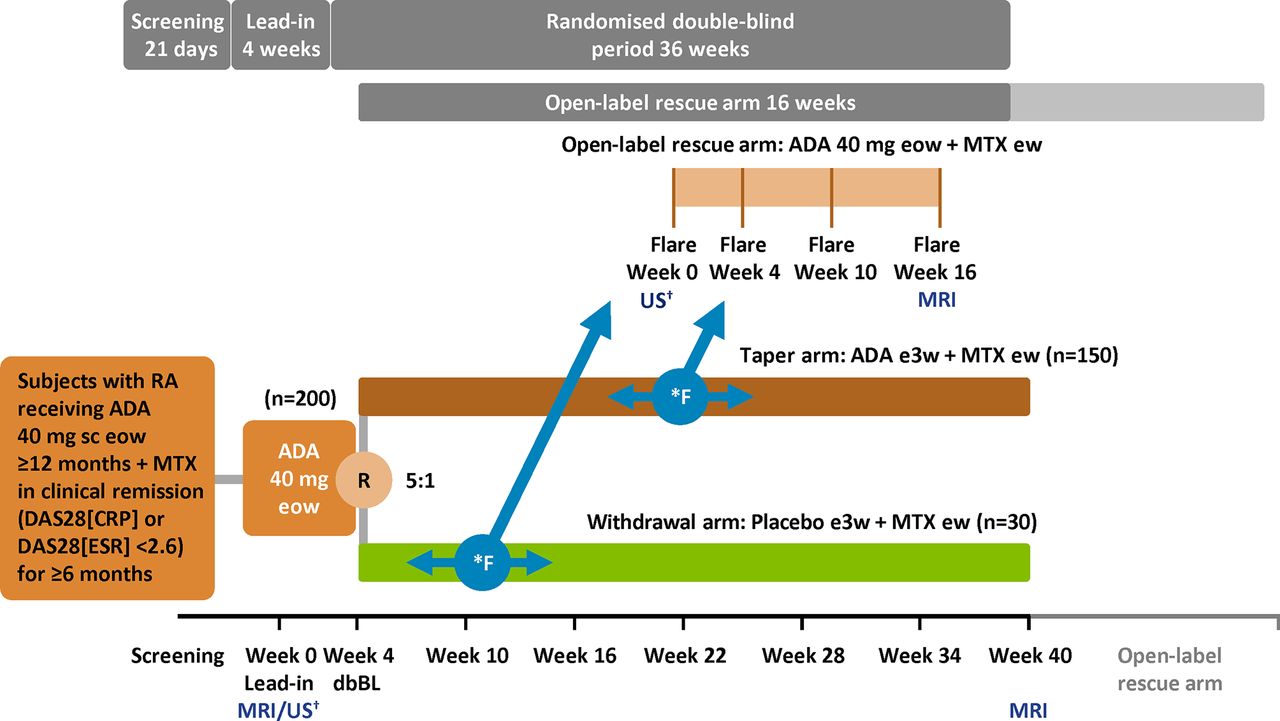{kind=link}
Study design. *Flare at any time point; flare defined as either (1) DAS28(ESR) ≥2.6 with an increase in DAS28(ESR) by >0.6 or (2) an increase in DAS28(ESR) by ≥1.2 from dbBL irrespective of absolute DAS28(ESR) score. Participants who flare at any time during the randomised double-blind period will be switched to open-label ADA 40 mg eow and continue in the open-label rescue arm for 16 weeks up to a maximum study duration of 56 weeks. †If applicable. ADA, adalimumab; DAS28(CRP), Disease Activity Score based on 28 joints and C reactive protein; DAS28(ESR), Disease Activity Score based on 28 joints and erythrocyte sedimentation rate; dbBL, double-blind baseline; ew, every week; eow, every other week; e3w; every 3 weeks; F, flare; MTX, methotrexate; R, randomisation; RA, rheumatoid arthritis; sc, subcutaneously; US, ultrasound.
If a participant prematurely discontinues study drug use, a termination visit must be completed within 2 weeks of the last dose of study drug and preferably prior to the initiation of another therapy. After discontinuation, the participant will be treated in accordance with the investigator’s best clinical judgement. A follow-up phone call will be made to all participants approximately 70 days after the last dose of study drug to determine the status of any ongoing adverse events (AEs) or the occurrence of any new AEs. Information regarding incentives for participants and provisions for treating and/or compensating participants who are harmed as a consequence of participation in the study are listed in online supplementary patient consent form file.
Supplementary material 1
Blinding and treatment allocation
All study sponsor personnel with direct oversight of the conduct and management of the trial (with the exception of the sponsor Drug Supply Management Team), the investigator, study site personnel and the participants remain blinded to treatment throughout the double-blind period of the study. Participants are assigned a unique identification number by an interactive response technology (IRT) system at screening and are centrally randomised. The treatment group assignments are not provided to the site. The IRT system provides access to participant treatment information in the case of medical emergency.
Objectives
The overall aim of the PREDICTRA study is to generate data on participant and disease characteristics, including inflammatory markers detected by sensitive imaging techniques, other biomarkers and adalimumab drug levels that potentially predict the clinical course (ie, the occurrence of flares) of the adalimumab 40 mg every 3 week dose-tapering regimen. The primary objective is to investigate the association between residual inflammatory disease activity at lead-in baseline (as detected by MRI scores for synovitis and osteitis) and the occurrence of flares in participants with RA randomised to receive a dose-tapering regimen of adalimumab controlled by withdrawal of adalimumab.
Key secondary objectives include assessing the (1) occurrence and severity of flares and the time to flares in the taper and withdrawal arms; (2) association between double-blind baseline participant demographic and disease characteristics and the occurrence of flares; (3) association between double-blind baseline adalimumab trough concentrations and the occurrence of flares; (4) effectiveness of rescue therapy with open-label adalimumab 40 mg eow over 16 weeks in participants experiencing a flare; (5) inflammatory and structural changes from lead-in baseline to final visit in the taper, withdrawal and rescue arms (based on Rheumatoid Arthritis MRI Score (RAMRIS)); (6) associations between lead-in baseline MRI and flare in subgroups of participants who meet additional clinical remission criteria at double-blind baseline, including Simplified Disease Activity Index (SDAI) ≤3.3, Clinical Disease Activity Index (CDAI) ≤2.8 and ACR/EULAR 2011 Boolean-based remission; (7) course of disease and patient-reported outcome (PRO) measures in the taper, withdrawal and rescue arms overall and in the double-blind baseline subgroup and (8) incidence of participants with positive tests for anti-adalimumab antibodies (AAAs) in the taper and withdrawal arms throughout the study.
An US substudy will have the following exploratory objectives: to investigate the (1) association between double-blind baseline US scores and the occurrence of RA flares, (2) association between double-blind baseline US scores and lead-in baseline RAMRIS scores, (3) change in the US scores from double-blind baseline to the time of RA flare in the taper and withdrawal arms. A 46-joint (40 joint areas) validated assessment and 18-tendon/tendon compartment assessment will be performed using grayscale US (GSUS) and power Doppler US (PDUS). If the ultrasonographer has not participated in US evaluation in a multicentre study in RA, training in standardisation of the scanning method and synovitis scoring system will be provided.
As a separate exploratory objective, the association between different biomarkers and the occurrence of flares will be studied in participants who provide a separate dedicated consent. Optionally, participants may provide material for pharmacogenetic and RNA analyses.
Assessments and variables
Key assessments that will be performed during the study are listed in table 1. Overall, eight clinical domains of flare assessment are being evaluated: pain, function, swollen joints, tender joints, PGA, stiffness, fatigue and flare severity. Objective assessments include MRI, US, ESR, CRP, chemistry/haematology, rheumatoid factor, anticitrullinated peptide antibody, antinuclear antibodies and pharmacokinetics and immunogenicity (trough adalimumab concentrations and AAAs).
Key assessments in the PREDICTRA study
The primary explanatory efficacy variables include the lead-in baseline hand and wrist synovitis and osteitis (bone marrow oedema) RAMRIS scores as well as a composite of both scores; the dependent variable is the occurrence of flare up to week 40. Secondary efficacy variables include the following: time to flare; flare severity; proportion of participants with a flare; participant demographics and clinical disease characteristics at double-blind baseline; proportion of flared participants who regain clinical remission (defined as DAS28(ESR) <2.6 or DAS28(ESR) decrease of >1.2 if DAS28(ESR) was <2.6 at flare), proportion of participants with LDA (DAS28(ESR) <3.2) and time to regain remission in the rescue arm; change from double-blind baseline in DAS28(ESR), CDAI, SDAI and the Health Assessment Questionnaire Disability Index (HAQ-DI); proportion of participants maintaining clinical remission (defined as DAS28(ESR) <2.6, SDAI ≤3.3 or CDAI ≤2.8) throughout the study; change from lead-in baseline to week 40 or final visit in synovitis, bone marrow oedema and erosions RAMRIS scores; change from double-blind baseline in HAQ-DI scores over time; proportion of participants with HAQ-DI scores ≤0.5 at double-blind baseline and at week 40; change from double-blind baseline in Routine Assessment of Patient Index Data (RAPID 3) scores during visits; change from flare week 0 in RAPID 3 at-home assessments and change from double-blind baseline in swollen joint count (both 28 and 66 joints), tender joint count (both 28 and 68 joints), PGA of disease activity, PGA of pain, Physician Global Assessment of disease activity, morning stiffness assessment, sleep disturbance assessment, Treatment Satisfaction Questionnaire for Medication, Work Productivity and Activity Impairment, Short Form-36 Health Survey Questionnaire, Functional Assessment of Chronic Illness Therapy—fatigue, CRP levels and ESR.
Exploratory variables include change from double-blind baseline to the time of flare in GSUS individual scores for SH, tenosynovitis and erosions, PDUS individual scores for synovitis/vascularisation and in a composite score for synovitis as well as change in the following biomarkers: matrix metalloproteinase 3, type I collagen neo-epitope, type III collagen neo-epitope, MMP-degraded CRP, MMP-degraded citrullinated vimentin, serum amyloid-associated protein, interleukin-6, chemokine (C-X-C motif) ligand 10 (CXCL-10) and CXCL-13.
Safety
The occurrence of AEs, based on clinical and laboratory data, will be collected at each postscreening visit and any time during the study as well as for up to 70 days after discontinuation of study treatment.
Statistical methods
Approximately 200 participants will be enrolled into the lead-in period of the study. Accounting for a 10% discontinuation rate, the study is designed for approximately 180 participants to enter the double-blind period (150 participants randomised in the taper arm and 30 participants in the withdrawal arm) to address the primary objective. A sample size of 150 patients in the dose tapering group will ensure a precision for the estimation with the width of 90% CI no more than 0.03 for an OR 1.03, no more than 0.07 for an OR 1.1 and no more than 0.14 for an OR 1.2 for occurrence of flare with baseline MRI score. Such sample size will also ensure a precision for the estimation of a correlation coefficient ρ with the width of 90% CI of ρ no more than 0.26 for a mild correlation coefficient 0.28, no more than 0.18 for a moderate correlation coefficient 0.55 and no more than 0.13 for a higher correlation with ρ=0.67. Assuming a 30% flare rate, this sample size will provide the precision that the 2-sided 90% CI of the flare rate has a half width no more than 6%.
The analysis population for the assessment of efficacy and safety will include all participants who receive ≥1 dose of double-blind study treatment. All statistical inference will be based on a 2-sided alpha level of 0.1. No multiplicity adjustment will be conducted. The quality of the data will be ensured through manual and automated checks. The analysis will be performed using SAS (SAS Institute, Cary, North Carolina, USA).
For the primary objective, the main analysis will examine the association between the occurrence of flares and baseline RAMRIS scores in the taper arm using logistic regression. Each OR will be calculated with a 90% CI. For this analysis, missing flare status for participants who discontinued prematurely (ie, discontinued without detection of a flare) during the double-blind period will be imputed from the last DAS28(ESR) measurement; however, any participant who received rescue therapy will be considered to be flared. In addition, descriptive statistics of baseline RAMRIS scores will be provided for participants with and without a disease flare, and the between-group difference in the mean scores will be calculated with 90% CIs. Linear regression will be used to model the relationship between the DAS28(ESR) at flaring (or at the end of study for participants without a flare) and baseline RAMRIS scores. Receiver operating characteristic curves will be used to investigate the potential flare prediction criteria (ie, cut-off values) based on RAMRIS scores. All model-based analyses may be adjusted for double-blind baseline values of demographic and clinical participant characteristics (eg, age, disease duration, previous and concomitant treatment) when appropriate. Similar analyses will be conducted for RAMRIS synovitis scores, bone marrow oedema scores and the composite of both scores.
For the secondary objectives, the association between the specified double-blind baseline parameters and the occurrence of flares will be examined using similar analyses to those used for the assessment of the primary objective. In addition, the proportion of participants with a flare, the proportion within each level of flare severity and the proportion of flared participants who regain clinical remission will be calculated with 90% CIs. Time to flare will be summarised using Kaplan-Meier survival techniques. Change from baseline in the MRI scores and the specified clinical and health outcome measures will be summarised over time separately for flared and non-flared participants. The calculations will be performed in both the taper arm and the withdrawal arm.
For exploratory objectives, US scores (SH, tenosynovitis and erosions using GSUS; synovitis/vascularisation using PDUS; synovitis composite score) at double-blind baseline, at flare and for the change between the two times will be summarised for participants with and without flares. A similar analysis will be conducted for double-blind baseline biomarker values. In addition, a linear regression approach will be used to model the relation between the US scores and RAMRIS scores at double-blind baseline. Additional analyses will be performed if deemed necessary.
The number and proportion of participants experiencing treatment-emergent AEs (ie, events on or after the first dose of study medication through the post-treatment assessment period) will be summarised by treatment, system organ class and Medical Dictionary for Regulatory Activities preferred term. In addition, summary of treatment-emergent AEs by severity and relationship to study drug, as judged by the investigators, will be reported.
An interim analysis of baseline characteristics of the trial population is projected to occur in November 2017.
Ethics and dissemination
The study is conducted in accordance with the International Conference on Harmonisation guidelines, local laws and the ethical principles of the Declaration of Helsinki. All participants are required to sign a written informed consent statement before the start of any study procedures. The study sponsor abides by the Principles on Conduct of Clinical Trials and Communication of Clinical Trial results of the Pharmaceutical Research and Manufacturers of America and all relevant state and federal laws. Because this is a phase IV study of a well-established medicinal product, there is no Data Monitoring Committee. Each investigator maintains a confidential identification code list, not to be retrieved by AbbVie, of the participants that he or she has enrolled in the study. The AbbVie Quality Assurance team audits at least 10% of the study sites; the actual number of audits may be higher should the team deem it necessary to perform additional audits. A protocol amendment was submitted for approval to research ethics committees, institutional review boards and other applicable regulatory institutions. The registration at clinicaltrials.gov was updated per the amendment; investigators were notified globally by newsletter and email and via individual follow-up to obtain a signature acknowledging receipt of the amendment. The final results will be shared with all relevant parties and disseminated through peer-reviewed journals and/or scientific conferences.
Discussion
The PREDICTRA trial aims to assess the most comprehensive array of predictors, including imaging of patient outcome (flare) on dose tapering of a bDMARD. The study was not designed to assess a treatment effect, but rather to predict which patients may undergo dose tapering of adalimumab with maintenance of RA disease remission.
The strengths of the PREDICTRA study include the use of MRI as a sensitive imaging technique, in addition to US, compared with previous trials. US does not detect bone marrow oedema, which, along with synovitis, predicts structural progression.30 Although ultrasonography is a highly feasible tool for diagnosis and monitoring of patients with RA in the clinic, MRI is reliable and highly responsive when used (eg, to detect subclinical inflammation (synovitis and osteitis)31,32) in multisite clinical studies. Therefore, the combined use of MRI and US in PREDICTRA allows for a more comprehensive evaluation of musculoskeletal inflammation.
The randomised, double-blind, placebo-controlled study design reduces bias by eliminating expectations of treatment benefit or its loss based on treatment assignment. The presence of a control withdrawal arm comprising one-sixth of the participants is also expected to encourage all participants, because of uncertainty about whether they are receiving adalimumab or not, to sensitively monitor their own symptoms through the PRO of PGA (a component of DAS28), which contributes to the definition of flare. The open-label rescue arm of up to 16 weeks of adalimumab therapy at 40 mg eow provides an opportunity to assess the effectiveness of retreatment with the standard adalimumab regimen. The use of MRI will also enable sensitive measurement of structural progression in the adalimumab taper, withdrawal and rescue arms relative to disease control. A limitation of the study design of PREDICTRA is that the duration of the trial may be insufficient to assess long-term progression of MRI-detected structural joint damage. Another limitation is multiple testing bias when investigating several possible predictors. Furthermore, some measurements are costly or somewhat difficult to routinely do in an outpatient clinic. Finally, because a validated consensus definition of RA flare has not yet been established,33 the ability to compare the results from PREDICTRA with findings from similar studies may be limited.
Study enrolment began in December 2014 and was closed in July 2017. Final results will be available in 2019.
Acknowledgments
The authors thank Anabela Cardoso, MD, formerly of AbbVie, for her contributions to the study. Medical writing assistance was provided by Maria Hovenden, PhD and Michael J Theisen, PhD of Complete Publication Solutions and was supported by AbbVie. AbbVie and the authors thank the participants in the clinical trial and all study investigators for their contributions.
References
Footnotes
Contributors Study concept and design: PE, GRB, EN, YZ, PGC. Protocol and statistical analysis plan development: PE, GRB, EN, YZ, MH, PGC. Drafting of the manuscript and critical revision of the manuscript for important intellectual content: All authors.
Funding AbbVie (North Chicago, Illinois, USA) funded this ongoing study (EudraCT 2014-001114-26 and NCT02198651), contributed to the design and was involved in the collection, analysis and interpretation of the data and in the writing, review and approval of this publication. PE and PGC are supported in part by the National Institute for Health Research (NIHR) Leeds Biomedical Research Centre.
Disclaimer The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health.
Competing interests PE has received research grants and/or consulting fees from AbbVie, Bristol-Myers Squibb, Lilly, Merck, Novartis, Pfizer, Roche, Sandoz and UCB. GRB has received research grants and/or consulting fees from AbbVie, Bristol-Myers Squibb, Lilly, MSD, Novartis, Pfizer, Roche, Sandoz and UCB. EN has received speaker fees from AbbVie, Roche, Bristol-Myers Squibb, Pfizer, UCB, Lilly, Novartis, Janssen and Celgene GmbH and honoraria from AbbVie. YZ and MH are full-time employees of AbbVie and may hold AbbVie stock or stock options. PGC has received speakers’ bureau or consulting fees from AbbVie, Bristol- Myers Squibb, Lilly, Novartis, Pfizer and Roche.
Patient consent Not required.
Ethics approval The study protocol was approved by an independent ethics committee or institutional review board at each study site.
Provenance and peer review Not commissioned; externally peer reviewed.