

Abstract
Objective. To determine survival and causes of death in an unselected and complete cohort of Norwegian patients with systemic sclerosis (SSc) compared to the background population.
Methods. Multiple methods were used to identify every patient with SSc living in southeast Norway, with a denominator population of 2,707,012, between 1999 and 2009. All patients who met either the American College of Rheumatology criteria or the Medsger and LeRoy criteria for SSc were included. Every patient was matched for sex and age with 15 healthy controls drawn from the national population registry. Vital status at January 1, 2010, was provided for patients and controls by the national population registry. Causes of death were obtained from death certificates and by chart review.
Results. Forty-three (14%) of 312 patients with SSc died during the study period. The standardized mortality rate (SMR) was estimated to be 2.03 for the entire cohort and 5.33 for the subgroup with diffuse cutaneous (dc) SSc. The 5- and 10-year survival rates were 91% and 70%, respectively, for dcSSc and 98% and 93% for limited cutaneous (lc) SSc. Causes of death were related to SSc in 24/43 (56%) patients, mostly cardiopulmonary diseases (n = 13), including pulmonary hypertension (n = 8). Factors associated with fatal outcome included male sex, dcSSc, pulmonary hypertension, and interstitial lung disease.
Conclusion. Compared to the Norwegian background population, our cohort of 312 unselected patients with SSc had decreased survival. The survival rates observed were, however, better than those previously reported from SSc referral centers.
Systemic sclerosis (SSc) is a serious multiorgan disease characterized by progressive vasculopathy, fibrosis, and distinct serum autoantibodies1,2. The etiology of SSc is largely unknown, but involves both genetic and environmental factors3. Classification of SSc is still done with the American College of Rheumatology (ACR) classification criteria from 19804, but alternative classification criteria are increasingly used, particularly the Medsger and LeRoy system, which divides patients into 3 subsets: diffuse cutaneous SSc (dcSSc), limited cutaneous SSc (lcSSc), and limited SSc (lSSc)5. SSc incidence data vary from 0.6 to 19/100,000 and prevalence rates range from 5/100,000 to 30/100,0006. Recently, we estimated the prevalence of SSc in Norway at 9.9/100,0007.
SSc is clearly a disorder with increased mortality, but the observed mortality rates differ considerably between studies. Reported standardized mortality ratios (SMR) range from 1.5 to 7.28,9,10,11,12,13 and the 5- and 10-year cumulative survival estimates range from 80% to 90% and 60% to 85%, respectively13,14,15,16,17,18. Moreover, the frequency of deaths related to SSc varies from 30% to 80%13,19,20,21,22,23,24. Because most previous mortality studies in SSc were conducted in single centers with selected cohorts, the large variations may partly be explained by selection bias. Also, previous studies used different SSc criteria and life tables, rather than disease control groups, to determine the SMR. Notwithstanding, patients with SSc continue to carry one of the highest risks of mortality of all connective tissue diseases24,25.
Factors previously associated with increased mortality have included male sex, older age at disease onset, dcSSc subtype, and the presence of serum antitopoisomerase I antibodies (ATA) as well as cardiovascular, pulmonary, and renal disease involvement14,24,26,27,28,29,30,31,32,33,34,35.
Norway is particularly suitable for mortality studies because of reliable mortality statistics, which are necessary to evaluate the health effects of diseases at the population level. We recently established an unselected SSc cohort, which includes every identifiable patient in southeast Norway (population 2.7 million). The patients were included by the ACR and Medsger and Leroy criteria and were followed for a mean 11 years with no loss to followup. Using the unique possibility of comparing patients with controls matched for sex/age from the national population registry, we were able to obtain complete SMR data. We describe the survival and causes of death of unselected patients with SSc in southeast Norway compared to the Norwegian background population.
MATERIALS AND METHODS
Study cohort
The study cohort, which included every identifiable patient with SSc living in the study area between January 1, 1999, and December 31, 2009, has been described7. Briefly, 5 different acquisition routes were used as follows to identify all patients in the study area who had been registered with an SSc diagnosis code (i.e., M34) according to the 10th revised version of the International Classification of Diseases (ICD-10), which has been used in all Norwegian hospitals since 1999. (1) The Norwegian Systemic Connective Tissue Disease And Vasculitis Registry at the Department of Rheumatology, Oslo University Hospital (OUH) was searched. (2) All the other 7 departments of rheumatology in southeast Norway were asked to search their hospital databases. (3) All rheumatology consultants practicing within the study region were invited to provide details of patients under their care whom they considered to have SSc. (4) The 1 rheumatology department for children in Norway (located at OUH) was asked to search its database for juvenile patients with SSc. (5) The Department of Dermatology at the OUH (the only dermatology department in southeast Norway) was asked to search its database for SSc patients not taken care of by the rheumatology department. Because every person living in Norway has a unique Norwegian identity number, no patients were double-registered.
The study was approved by the regional committee of health and medical research ethics in southeast Norway.
Disease characteristics
All the 312 patients fulfilled either the ACR criteria or the Medsger and LeRoy criteria for SSc4,5. Patients were divided into 3 subgroups: (1) lSSc with Raynaud phenomenon (RP) plus SSc-type nailfold capillary pattern or SSc selective autoantibodies; (2) lcSSc with skin thickening distal to the elbow/knee; and (3) dcSSc7,24.
Disease onset was defined as the timepoint when the first non-RP symptom was reported and was retrospectively registered from 1965 until study end. The observation period was terminated by the end of 2009 or at the time of death. Disease duration was defined as the time from onset of the first non-RP symptom to the end of the observation period or to time of death. The period from first non-RP symptom to time of SSc diagnosis was defined as diagnostic delay. The period from the SSc diagnosis until study end or death was defined as the followup time.
Clinical characteristics
The presence of the following disease measures during the disease course was recorded retrospectively by chart review: (1) serum autoantibody status: antinuclear antibody (ANA), anticentromere antibody (ACA), and ATA; (2) digital ulcers (DU); (3) scleroderma renal crisis (SRC); (4) gastric antral vascular ectasia (GAVE) as evidenced by gastroscopy; (5) the presence of interstitial lung disease (ILD) as evidenced by high-resolution computed tomography (HRCT) of the lungs and pulmonary function tests (PFT); and (6) pulmonary hypertension (PH). The presence of PH was defined according to the latest criteria from the European Society of Cardiology as mean pulmonary artery pressure (PAP) ≥ 25 mm HG by right heart catheterization (RHC) at rest36. RHC was conducted when PH was suspected clinically and/or by elevated systolic PAP by echocardiography. All patients who underwent RHC were evaluated by an experienced cardiologist to exclude that the PH was secondary to heart disease.
In accord with recent PH criteria37, the PH was classified as ILD-associated (PH-ILD) if the patient had a restrictive PFT pattern with forced vital capacity (FVC) < 70% and severe lung fibrosis on HRCT of the lungs, while patients who had FVC > 70% or no/mild HRCT abnormalities were classified as having pulmonary arterial hypertension (PAH). ILD was staged as mild or extensive according to the criteria suggested by Goh, et al38. We had access to charts on all 312 patients, but not all the charts had complete data on every clinical variable. In the statistical analyses, missing data were recorded as “parameter not present.” To assess the influence of clinical variables on mortality, OR were calculated. Patients’ treatment was not evaluated.
Assessment of SMR and survival rates
Using the personal identification numbers, we were able to identify every death and all the reported causes of death in the SSc cohort between January 1, 1999, and January 1, 2010. Vital status at the end of the study was established by contacting the Norwegian Central Person Register and by chart review. To calculate the SMR, Statistics Norway provided a control group from the general Norwegian population. Altogether, 15 random controls were selected for each individual patient with SSc. The controls were matched with the SSc patients for sex, year of birth, and residence area. Additionally, all 15 controls were alive on the date of patient’s disease onset. For calculation of SMR in the followup period we excluded all controls dying during the period of diagnostic delay and reduced the number of controls per patient accordingly. Cumulative survival rates were computed by the Kaplan-Meier method and significance was tested with the log-rank test. Survival curves were compared to curves of the control group.
Causes of death
Causes of death were based on information from medical charts (38 patients), death certificates (all 43 deceased patients), and autopsy (1 patient). When the information from the different sources was not consistent (4 patients), we used data from the medical chart and not from the death certificate. On 3 death certificates, SSc was not mentioned as a contributing cause of death (cause of death unknown). Causes of death of the control group were given by Statistics Norway. The causes of death of SSc patients were classified as SSc-related mortality and mortality unrelated to SSc. SSc-related deaths were defined as deaths related to progressive, active SSc or deaths caused by organ failure directly related to SSc. Deaths unrelated to SSc were of all other causes and divided into several subgroups. If there was no notification on the death certificate or in the patient chart, the cause of death was defined as “unknown.”
Statistics
All information was imported to the Microsoft Office Access database after scanning with Cardiff Teleform 10.1 software. Statistical analyses were performed by SPSS, version 18. Causes of death were analyzed using standard descriptive statistics. Kaplan-Meier survival curves and significance were tested with the log-rank test. Items with significant effects on survival were entered into the Cox proportional hazards model. The SMR was calculated as the ratio between the observed rate of death in the SSc cohort and the observed rate of death in the comparable age- and sex-matched control group from Statistics Norway, representing the Norwegian background population. The association between potential risk factors and mortality was quantified by OR with 95% CI. Further, all independent risk factors significantly associated with SSc from the univariate analysis were considered as candidates in the multivariate logistic regression analysis. In the univariate analysis we considered the following potential risk factors: sex, age, disease subtype, ANA, ACA, ATA, GAVE, DU, SRC, ILD, and PAH. A manual backward stepwise elimination procedure using a multivariate logistic regression model was performed to identify independent risk factors of mortality. Multivariate analyses were preceded by estimation of correlation between risk factors. Differences in clinical findings between survivors and deceased patients were analyzed using Pearson chi-square test for contingency tables for categorical variables and independent sample t test for continuous variables.
RESULTS
Demographic and clinical variables
The total study cohort consisted of 95% whites and included 312 patients who fulfilled the ACR classification criteria for SSc and/or the Medsger and LeRoy criteria (Table 1). Forty-three (14%) of these 312 patients died during the study period from December 31, 1999, until January 1, 2010. The deceased patient group had a higher mean age at disease onset and higher mean age at diagnosis and at death, but the same mean disease duration as the group of surviving patients (Table 1). The mean diagnostic delay was 2.9 years (SD 4.3). The deceased patient group consisted of 6 men with lcSSc and 9 with dcSSc compared to 20 women with lcSSc and 8 with dcSSc. Men with dcSSc had a higher mortality (43%) compared to women with dcSSc (17%) and to men and women with lcSSc (12% and 10%, respectively). In the small subgroup with lSSc, only 1 death was observed during the study period.
Demographic data and disease subtype in the study cohort. Data are mean (SD) unless otherwise indicated.
Chart review showed a higher frequency of ILD, PH associated with ILD (PH-ILD), and PAH in the 43 patients who died during the study period than in the surviving patients (Figure 1). Indeed, univariate analyses identified ILD and PH (PAH and PH-ILD) as factors associated with mortality (Table 2). Male sex and the dcSSc subtype were also factors associated with a poor prognosis, whereas ACA was negatively associated with mortality (Table 2). The multivariate logistic regression analysis using a manual backward elimination procedure showed that PAH (OR 8.0, 95% CI 3.4–18.8) followed by ILD (OR 3.1, 95% CI 1.5–6.6) were the strongest predictors for mortality.
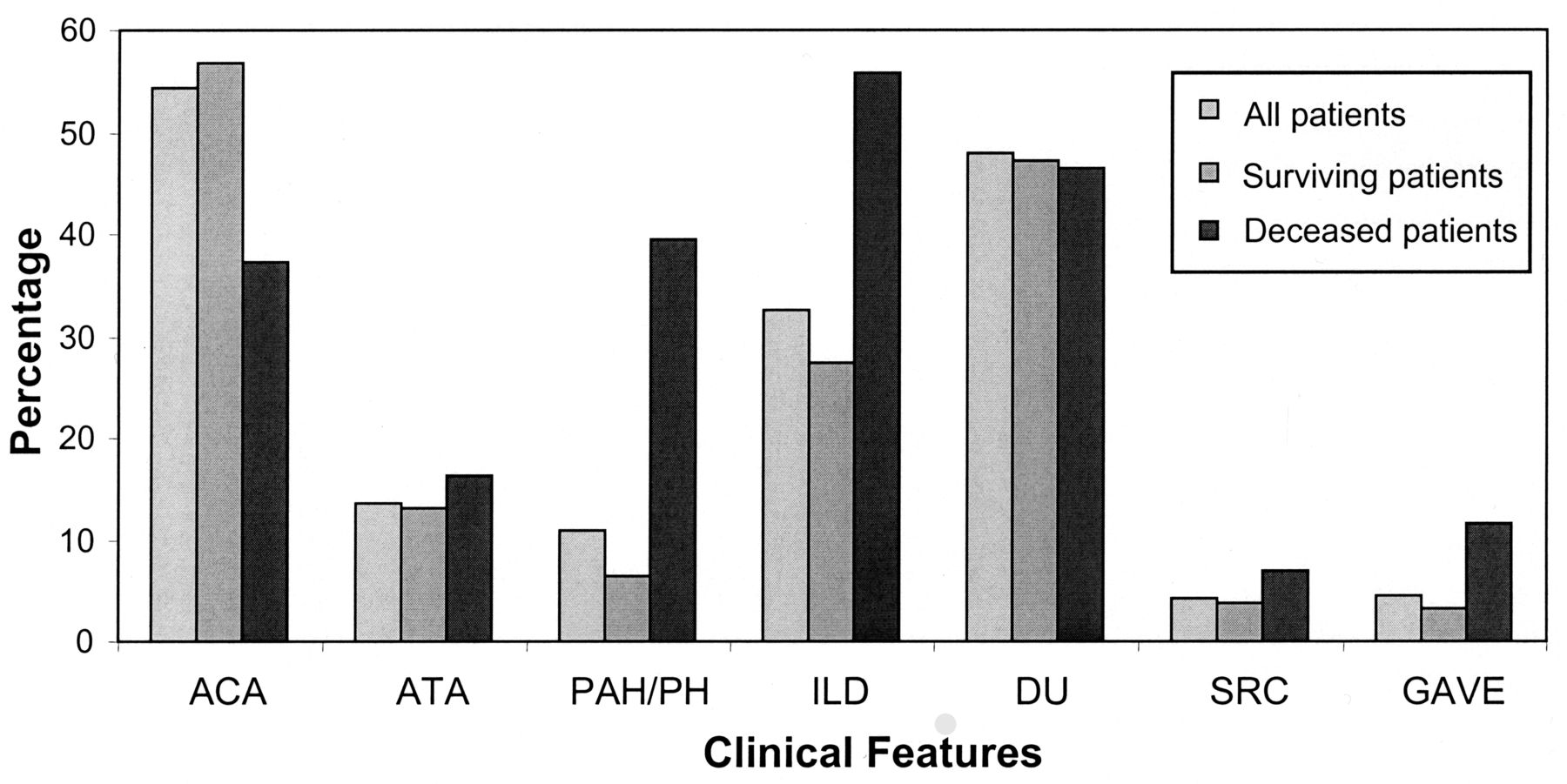
Clinical features and serum autoantibodies of 312 patients with systemic sclerosis: 269 surviving, 43 deceased. ACA: anticentromere antibody; ATA: antitopoisomerase 1 antibody; PAH: pulmonary arterial hypertension; ILD: interstitial lung disease; DU: digital ulcers; SRC: scleroderma renal crisis; GAVE: gastric antral vascular ectasia.
Overview of clinical findings in 269 living and 43 deceased patients and their relative prognostic value assessed by OR.
Standardized mortality ratios
During the observation period, the mortality rate in the patients was 14%, while it was 7% in the age- and sex-matched control group (i.e., 317 deaths in 4680 controls). Thus, the overall SMR of the observation period was 2.03. The all-cause SMR was higher in men than in women; SMR for dcSSc was 5.33 compared to 1.62 for lcSSc (Table 3). We also calculated the overall SMR of the followup period separately and found that this was 2.00 (95% CI 1.4–2.7), i.e., not different from the SMR of the total observation period.
Estimated risk of death for the patients with systemic sclerosis (SSc) compared with the control group.
Survival rates
The Kaplan-Meier cumulative 5-year overall survival for patients with SSc was 95%, and the 10-year survival was estimated to be 86% (Figure 2a). The 5- and 10-year survival rates for lcSSc were estimated to be 98% and 93%, and 91% and 70% for dcSSc, respectively (Figure 2b).

Survival of patients with systemic sclerosis (SSc). A. Overall survival of SSc patients compared to the Norwegian background population. B. Survival of patients with dcSSc and lcSSc. dcSSc: diffuse cutaneous SSc; lcSSc: limited cutaneous SSc.
Causes of death
Chart review and review of death certificates suggested that the death was attributable to SSc in 24 of the 43 deceased patients (Table 4). Of the remaining 19 patients, 16 had causes of death not attributable to the disease and 3 died sudden deaths that, according to the death certificates, had unknown causes. Review of the medical charts of these 3 patients, all having clinically stable SSc, did not provide any further information on the causes of death.
Causes of death of the 43 deceased patients with systemic sclerosis (SSc).
In total, 56% of the SSc-attributable deaths were due to cardiopulmonary diseases, with pulmonary fibrosis (20%) and PAH/PH (20%) the most frequent causes (Table 4). All 5 patients dying of pulmonary fibrosis had extensive fibrosis according to the staging system of Goh, et al38. All patients dying of PAH/PH had precapillary PH; no signs of postcapillary PH were noted.
Another 25% of the patients died of SSc-associated gastrointestinal causes, such as bleeding, aspiration, cachexia, and malabsorption. Renal causes accounted for the death of 5 patients (20%). Three of the 5 patients with renal failure were diagnosed with SRC during their disease course (12.5% of SSc-related deaths). They all died within 3 years after their SRC diagnosis.
The 16 cases with non-SSc-related causes of death included malignancies (n = 6), infections (n = 2), coronary arterial disease (n = 2), obstructive lung diseases (n = 2), stroke (n = 2), and bleeding anemia (n = 2; Table 4).
Statistics Norway provided data on the causes of death of the control group, as stated on their death certificates. The most common causes of death in the controls were malignancies (45%), cardiovascular disease (20%), stroke (11%), infections (6%), and obstructive lung disease (6%). No comparison of the causes of death between patients with SSc and controls was performed.
DISCUSSION
Data on the survival and causes of death in unselected SSc cohorts are limited. We followed 312 patients with SSc over an 11-year period and compared the mortality in this SSc cohort with age- and sex-matched controls from the Norwegian background population. Our results reinforce the view that SSc is a disease with increased mortality and that male sex, dcSSc, and cardiopulmonary diseases are factors associated with a poor prognosis.
Both the observed SMR of 2.03 and the 5- and 10-year survival rates of 95% and 86% indicate that the mortality of SSc in southeast Norway is lower than reported in most other countries8,10,14,15,17,19,21,39. We tentatively suggest, however, that the relatively low mortality reflects that the current cohort is unselected and therefore includes proportionally more patients with mild and early disease than cohorts from referral centers. In accord with the recent metaanalysis by Komocsi, et al30, we observed that males and patients with dcSSc had the worst outcome.
Since previous mortality studies have not included the lSSc subset, this subgroup was excluded from the mortality rate analyses. Although the mortality in the lSSc group was very low (only 1/35 patients died), the inclusion of this subgroup would have influenced the total SMR only slightly.
In our study, 56% of deaths were related to SSc. This frequency of SSc-attributable deaths and the distribution of SSc-related causes of mortality are comparable to figures reported from the European League Against Rheumatism Scleroderma Trials and Research group database, Elhai and Komocsi and their coworkers’ metaanalyses, and others12,19,20,22,30,31. However, we report a lower frequency of death caused by cardiovascular disease and malignancies than some other studies10,13,14,22,27,39. This might be due to the unselected design of this cohort, different interpretations of causes of death, or other causes, including genetic or environmental variations. SRC accounted for 12% of the SSc-related deaths; this is in the same range as recent data from Brazil and supports the results of Steen and Medsger showing decreasing frequency of SRC as an SSc-related cause of death19,27. Interestingly, we did not identify any deaths caused by arrhythmia, but cannot exclude the possibility that this was the cause in the 3 patients with sudden deaths.
The selection of the initial timepoint from which survival was estimated could have affected the survival results. Like Bryan, et al39 we used the first non-RP symptom as the initial timepoint when estimating disease duration. However, this might lead to higher survival for patients than for the controls. Further, our results using non-RP symptom as the initial timepoint may be difficult to compare with those preferring diagnosis as the start of the followup11,13,14,15,16,19. Therefore, we additionally estimated the overall SMR from the time of diagnosis, which yielded an SMR of 2.00 versus 2.03 using time of first non-RP symptom. Notably, the choice of the initial timepoint did not significantly affect the SMR. Only 10 (3%) of the 317 deaths observed in the 4680 age- and sex-matched controls occurred during the diagnostic delay period; this mainly explains the lack of difference. The rather long followup period is another partial explanation.
This study has some major strengths. First and probably most important is that our study provides reliable mortality data on an SSc cohort with unselected patients. It is, in fact, the only mortality study from Northern Europe with unselected patients10,14,39. Second, we believe that the robust acquisition strategy enabled us to enroll (almost) all the patients with SSc in the study area and to establish a complete SSc cohort. Third, the composition of the SSc cohort, with 75% women and about 25% patients with dcSSc, is similar to population-based cohorts, but different from the selected cohorts reported in the majority of mortality studies40. Fourth, because of the unique identification numbers in Norway and the official mortality statistics, no patients were lost to followup. Finally, to our knowledge the study is the first SSc mortality investigation that has used age- and sex-matched controls instead of life-table analyses to determine SMR.
The most important limitation of our study is probably the “immortality bias,” which was unavoidable given the study design41. This bias potentially leads to an underestimation of the true mortality and is caused by the inclusion of longtime survivors in the study population (i.e., patients who were diagnosed before 1999 and survived throughout the whole study period). We did, however, find that the SMR in the patient subgroup diagnosed before 1999 (n = 118) was only slightly lower than the SMR of the entire cohort (1.7 vs 2.0). This argues that the immortality bias had no major influence on the results. Another potential limitation is the retrospective design. All data on the causes of death relied on medical records, which frequently lack documentation, and death certificates, which may also lack critical information. Finally, because we assessed the mortality rate of SSc in a rather homogeneous population of white descent, our results are not necessarily comparable to data from other populations.
Our data show that mortality in SSc remains high, particularly in the subset of patients with dcSSc and male sex. The major causes of death were PH and lung fibrosis.
Acknowledgment
The authors thank Cecilie Kaufmann, Department of Rheumatology, Buskerud Hospital, Drammen; Anne Julsrud Haugen, Department of Rheumatology, Østfold Hospital, Moss; Anne Noraas Bendvold, Department of Rheumatology, Sørlandet Hospital, Kristiansand; Olav Bjørneboe, Department of Rheumatology, Martina Hansens Hospital, Baerum; Sven Gøran Sidenvall, Department of Rheumatology, Innlandet Hospital, Kongsvinger; Anne Salberg, Department of Rheumatology, Innlandet Hospital Lillehammer; and Hans Christian Gulseth, Department of Rheumatology, Betanien Hospital, Skien, Norway, for providing access to patient data. We also thank Mari Vårdal, Department of Biostatistics, Oslo University Hospital, and Geir Mjøen, Department of Medicine, Diakonhjemmets Sykehus, Oslo, for providing help with statistics.
Footnotes
-
Supported by grants from the Norwegian Women’s Public Health Association and the Scandinavian Rheumatology Research Foundation.
- Accepted for publication February 20, 2013.








{kind=link}
{kind=link}