Article Text

Abstract
Background: It has been shown that expression of the potent angiogenic factor, vascular endothelial growth factor (VEGF), and its receptors, flt-1 (VEGFR-1) and KDR/Flk-1 (VEGFR-2), increased during the development of liver fibrosis.
Aims: To elucidate the in vivo role of interaction between VEGF and its receptors in liver fibrogenesis.
Methods: A model of CCl4 induced hepatic fibrosis was used to assess the role of VEGFR-1 and VEGFR-2 by means of specific neutralising monoclonal antibodies (R-1mAb and R-2mAb, respectively). R-1mAb and R-2mAb were administered after two weeks of treatment with CCl4, and indices of fibrosis were assessed at eight weeks.
Results: Hepatic VEGF mRNA expression significantly increased during the development of liver fibrosis. Both R-1mAb and R-2mAb treatments significantly attenuated the development of fibrosis associated with suppression of neovascularisation in the liver. Hepatic hydroxyproline and serum fibrosis markers were also suppressed. Furthermore, the number of α-smooth muscle actin positive cells and α1(I)-procollagen mRNA expression were significantly suppressed by R-1mAb and R-2mAb treatment. The inhibitory effect of R-2mAb was more potent than that of R-1mAb, and combination treatment with both mAbs almost completely attenuated fibrosis development. Our in vitro study showed that VEGF treatment significantly stimulated proliferation of both activated hepatic stellate cells (HSC) and sinusoidal endothelial cells (SEC). VEGF also significantly increased α1(I)-procollagen mRNA expression in activated HSC.
Conclusions: These results suggest that the interaction of VEGF and its receptor, which reflected the combined effects of both on HSC and SEC, was a prerequisite for liver fibrosis development.
- vascular endothelial growth factor
- liver fibrosis
- angiogenesis
- hepatic stellate cells
- α-SMA, α smooth muscle actin
- ALT, aminotransferase aspartate
- A-M, Azan-Mallory
- ECM, extracellular matrix
- EC, endothelial cells
- HSC, hepatic stellate cells
- IgG, immunogloblin G
- mAb, neutralising monoclonal antibody
- PCR, polymerase chain reaction
- P-III-P, procollagen III-N-peptide
- R-1mAb, VEGFR-1 monoclonal antibody
- R-2mAb, VEGFR-2 monoclonal antibody
- SEC, hepatic sinusoidal endothelial cells
- VEGF, vascular endothelial growth factor
- VEGFR-1, fms-like tyrosine kinase (flt-1)
- VEGFR-2, kinase-insert domain-containing receptor/fetal liver kinase-1 (KDR/Flk-1)
- vWF, von Willebrand factor
Statistics from Altmetric.com
- α-SMA, α smooth muscle actin
- ALT, aminotransferase aspartate
- A-M, Azan-Mallory
- ECM, extracellular matrix
- EC, endothelial cells
- HSC, hepatic stellate cells
- IgG, immunogloblin G
- mAb, neutralising monoclonal antibody
- PCR, polymerase chain reaction
- P-III-P, procollagen III-N-peptide
- R-1mAb, VEGFR-1 monoclonal antibody
- R-2mAb, VEGFR-2 monoclonal antibody
- SEC, hepatic sinusoidal endothelial cells
- VEGF, vascular endothelial growth factor
- VEGFR-1, fms-like tyrosine kinase (flt-1)
- VEGFR-2, kinase-insert domain-containing receptor/fetal liver kinase-1 (KDR/Flk-1)
- vWF, von Willebrand factor
It is widely recognised that hepatic fibrosis development is associated with progression of chronic liver disease.1 It has been shown that capillarisation and phenotypic changes of the hepatic sinusoidal endothelial cells (SEC) occur during liver fibrosis development.2–4 The hepatic SEC are unique endothelial cells (EC) that show fenestration and lack of basement membrane. They reside both morphologically and functionally apart from the ordinary EC.5 Sinusoidal capillarisation involves changes in SEC, including loss of fenestration and deposition of a basement membrane, as well as alterations of the cell-cell and cell-matrix interactions in hepatic sinusoids.2–6 This so called capillarisation of SEC results either from development of neovessels or from alteration of the pre-existing sinusoids, both events possibly induced in response to angiogenic stimulation.6
Angiogenesis is the development of new vasculature from pre-existing blood vessels and/or circulating EC stem cells.7,8 Emerging evidence has shown that angiogenesis plays a pivotal role in many physiological and pathological processes, such as tumour growth, arthritis, psoriasis, and diabetic retinopathy.7,9 Although previous studies conducted to determine the molecular process associated with fibrosis and angiogenesis were performed independently, recent studies have revealed that both biological phenomena emerged synergistically.6 It was shown that neovascularisation significantly increased during the development of liver fibrosis in both human and animal experimental studies.10–12 Furthermore, a semisynthetic analogue of fumagilin, TNP-470, which possesses antiangiogenic activity, suppressed experimental liver fibrosis development.13 These results suggest that angiogenesis also plays an important role in the development of liver fibrosis.
Angiogenesis is regulated by the net balance between proangiogenic factors and angiogenic inhibitors. To date, many positive and negative angiogenic modulating factors have been identified. Among these, vascular endothelial growth factor (VEGF), also known as vascular permeability factor, is the most potent in the angiogenesis process.14–16 Emerging evidence has shown that VEGF plays a pivotal role in many cases of physiological and pathological angiogenesis. VEGF is not only an angiogenic factor, it is also known as a survival factor of EC.17–19 Regarding liver fibrosis, it has been shown recently that VEGF expression significantly increased during the course of liver fibrosis development in experimental studies, and that VEGF participated in sinusoidal capillarisation in the liver.11,12,20 In addition to hepatocytes, activated hepatic stellate cells (HSC), which play an important role in liver fibrogenesis, have been shown to increase VEGF expression during activation.21–24
Two tyrosine kinases, fms-like tyrosine kinase (flt-1: VEGFR-1) and the kinase insert domain-containing receptor/murine homologue, fetal liver kinase-1 (KDR/Flk-1: VEGFR-2), both of which are type III tyrosine kinase receptors, have been identified as the main VEGF receptors. By binding with high affinity to these two receptors, VEGF can stimulate EC proliferation, migration, and differentiation, and can induce angiogenesis in vitro and in vivo.16,25 It has been shown that these two receptors serve different biological roles in many pathological events.26–28 It has been reported that VEGFR-2 plays a more important role both in vitro and in vivo.15,16,29 However, recent studies have revealed that VEGFR-1 also plays certain roles in pathological angiogenesis, such as tumour growth.30–34 It has been shown that expression of VEGFR-1 and VEGFR-2 was induced during activation of HSC in vitro, although the upregulation patterns were different under different culture conditions, such as hypoxia and CCl4 treatment.22–24 In experimental liver fibrogenesis, it has been reported that VEGFR-1 expression increased in the liver, and VEGFR-2 was constitutively highly expressed although its expression level was not significantly altered.11,23 The in vivo role of the interaction between VEGF and its receptor in liver fibrosis development has not yet been elucidated.
In the present study, using the specific neutralising monoclonal antibodies of VEGFR-1 and VEGFR-2, we examined the biological role of VEGF and its receptors in the progression of liver fibrosis.
METHODS
Animals
Male BALB/c mice, aged six weeks, were purchased from Japan SLC Inc. (Hamamatsu, Shizuoka, Japan). They were housed in stainless steel mesh cages under controlled conditions (temperature 23±3°C and relative humidity 50±20%), with 10–15 air changes per hour and light illumination for 12 hours a day. The animals were allowed access to food and tap water ad libitum throughout the acclimatisation and experimental periods.
Compounds and animal treatment
Anti-VEGFR-1 and VEGFR-2 specific neutralising antibodies (R-1mAb and R-2mAb, respectively) were generated as described previously.31,33–36 Briefly, these antibodies were produced under large scale culture conditions in serum free media. The monoclonal antibodies (mAbs) were purified from conditioned media by affinity chromatography on a Gammabind-G-Sepharose column (Pharmacia Biotech, Piscataway, New Jersey, USA). The purity of the respective receptors was >99%, as determined by sodium dodecyl sulphate-polyacrylamide gel electrophoresis, and were verified to be free of endotoxin (<1 EU/ml) using a limulus amoebocyte lysate endotoxin detection kit (Pyrogen Tplus, Bio-Whittaker, Walkersville, Maryland, USA). It has been shown that R-2mAb exerts a VEGFR-2 inhibitory effect in a dose dependent manner, and that the maximal effect is achieved at a dose of 800 μg/mouse administered twice a week.34,37 We thus employed this dose in the current study.
Mice were divided into four groups (n=10 in each group). All experimental groups received CCl4 (2 ml/kg/body weight dissolved in 150 μl of corn oil) twice a week to develop liver fibrosis. After two weeks of treatment with CCl4, R-1mAb and R-2mAb (800 μg/mouse) were administered intraperitoneally to group 2 (G2) and group 3 (G3) twice a week on days different from those on which CCl4 was injected, respectively. In group 4 (G4), both R-1mAb and R-2mAb were administered simultaneously. Animals in group 1 (G1) received the same amount of control immunogloblin G (IgG) as described previously.35,36 Mice which received only corn oil were examined as a negative control group. After eight weeks of treatment with CCl4, all mice were killed under anaesthesia. All animal procedures were performed according to approved protocols and in accordance with the standard recommendations for the proper care and use of laboratory animals.
Histological and immunohistochemical examinations
In all experimental groups, 5 μm thick sections of formalin fixed and paraffin embedded livers were processed routinely for Azan-Mallory (A-M) staining for determination of liver fibrosis development. Immunohistochemical staining of α smooth muscle actin (α-SMA) was performed as previously described using paraffin embedded sections with a primary anti-α-SMA antibody (Dako, Kyoto, Japan).38–40 Semiquantitative analyses of fibrosis development and the immunopositive cell area were carried out with the Fuji-BAS 2000 image analysing system (Fuji, Tokyo, Japan) in six ocular fields (40× magnification) per specimen from five mice. We did not count α-SMA positive vessels in the portal area which were assumed to be hepatic arteries. We only included α-SMA positive cells in the sinusoidal lining for image analysis.38–40
Hepatic hydroxyproline content and serum markers
Hepatic hydroxyproline content was determined as previously described with 200 mg of frozen samples.40 The hydroxyproline content was expressed as μg/g wet liver. Alanine aminotransferase aspartate (ALT) and total bilirubin were assessed using routine laboratory methods. Serum hyaluronic acid and procollagen III-N-peptide (P-III-P) were also measured as described previously.38
Immunoprecipitation
To determine whether R-1mAb and R-2mAb at a dose of 800 μg/mouse suppressed autophosphorylation of the respective receptors in the liver, immunoprecipitation was performed as previously described35 Fifteen minutes after R-1mAb and R-2mAb were injected intraperitoneally, the liver was resected from three mice in each group and snap frozen immediately. The liver pool lysate solution was concentrated and used for immunoprecipitation. To conduct immunoprecipitation, liver lysates were immunoprecipitated with antiphosphotyrosine before conducting sodium dodecyl sulphate-polyacrylamide gel electrophoresis. Antityrosine (4G10) was purchased from Upstate Biotechnology (New York, USA) and anti-VEGFR-2 (C-1158) and VEGFR-1 (C-17) were obtained from Santa-Cruz (California, USA). Before western blotting, we stained each membrane with Ponceau solution (Sigma, Michigan, USA) to confirm that the same amounts of protein were immunoprecipitated (data not shown). The blots were developed using an amplified alkaline phosphatase immunoblot assay kit (Bio-Rad, California, USA).
RNA expression of VEGF, CD-31, and α1-(I)-procollagen by real time polymerase chain reaction
The VEGF, CD-31, which is used widely as a marker for neovascularisation, and α1-(I)-procollagen mRNA expression were evaluated by real time polymerase chain reaction (PCR), as described previously.38,41 mRNA was extracted from the whole liver of the animals in each experimental group (n=5). For cDNA synthesis, Taqman reverse transcription reagents were used as described in the manufacturer’s manual of the ABI Prism 7700 Sequence Detection System (PE Applied Biosystems, Foster City, California, USA), which was used for real time PCR amplification following the Taqman Universal PCR Master Mix Protocol (PE Applied Biosystems). Relative quantitation of gene expression was performed as described in the manual, using glyceraldehyde-3-phosphate dehydrogenase as an internal control. The threshold cycle and standard curve method were used for calculating the relative amount of the target RNA, as described for PE. The following temperatures were employed: hold at 50°C for two minutes, hold at 60°C for 30 minutes, hold at 94°C for five minutes, cycle 45 repeats at 94°C for one minute, at 55°C for one minute, and at 72°C for one minute. To prevent genomic DNA contamination, all RNA samples were subjected to DNase I digestion and checked by 40 cycles of PCR to confirm the absence of amplified DNA.
Isolation and culture of HSC and SEC
R-1mAb and R-2mAb only react with mice receptors and not with those of other species, such as the rat.31,42 Although we attempted several times to isolate pure HSC from the liver of mice, contamination with other types of non-parenchymal cells, such as EC, could not be ruled out. Furthermore, the yield of purified HSC from mice was too low to perform several experiments, as described previously.38 We thus employed HSC from the liver of the rat, and examined the effect of VEGF treatment on VEGF-receptor interaction in activated HSC. Liver HSC were isolated from the liver of F344 rats, as described previously,38 with a minor modification. Briefly, the liver was perfused with Krebs-Ringer solution followed by 0.1% pronase E and 0.032% collagenase (Nakarai, Kyoto, Japan) solution at 37°C. The digested liver was cut, minced, and incubated in Krebs-Ringer solution containing 0.08% pronase E, 0.04% collagenase, and 20 μl/ml DNase for 30 minutes at 37°C (pH 7.3). After passage through a nylon mesh, cells were centrifuged at 450 g for eight minutes. The HSC enriched fraction was obtained by centrifugation in 8.2% Nycodenz (Nycomed Pharma AS, Oslo, Norway) solution at 1400 g for 20 minutes. HSC in the upper white layer were washed by centrifugation at 450 g for eight minutes and suspended in DMEM medium containing 10% fetal calf serum, 100 U/ ml penicillin, and 100 mU/ml streptomycin. Cell viability was over 95%, as determined by the Trypan blue exclusion test. Cells were plated at a density of 5×105 cells/ml on either collagen I, an established model of culture activation,43 or basement membrane-like EHS matrix for six days in the presence of 20% fetal calf serum, serum starved for 48 hours, and treated with doses of 10 and 100 ng/ml of human recombinant VEGF (R&D systems Inc. Minneapolis, Minnesota, USA), as described previously.24 SEC were isolated from the rat liver with collagenase followed by differential centrifugation, as described previously,44 and grown in HuMedia-EG2 medium (Kurabo, Osaka, Japan) on a collagen I coated dish supplied with human recombinant VEGF (10 ng/ml) as VEGF is a prerequisite for SEC survival.45
In vitro proliferation assay
The effects of VEGF on in vitro proliferation of activated HSC and SEC were determined using the MTT assay, as described previously.46 Briefly, cell proliferation was quantified via conversion of tetrazolium, 3-(-4,5-diethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) by cells cultured in 96 well plates. Absorbance with a 540 nm filter represents conversion to formazan, which is directly proportional to the number of living cells. Absorbance was read with an ELISA plate recorder (n=6 per group).
Statistical analysis
To assess the statistical significance of intergroup differences in quantitative data, the Mann-Whitney U test was used to compare mean values between the two groups. The Kruskal-Wallis test was used to compare mean values between more than two groups.
RESULTS
VEGF mRNA expression
Firstly, we examined VEGF mRNA expression during CCl4 induced liver fibrosis development. Similar to previous reports,11,12 hepatic VEGF expression significantly increased during liver fibrosis development after CCl4 treatment. Neither R-1mAb nor R-2mAb treatment altered VEGF gene expression during development of fibrosis (fig 1). We performed a preliminary routine reverse transcription PCR, and found that among the alternative splicing mouse VEGF genes, VEGF164 and VEGF120 were abundant in the CCl4 treated liver (data not shown).

Vascular endothelial growth factor (VEGF) mRNA expression in the CCl4 treated liver. VEGF mRNA expression was examined by real time polymerase chain reaction as described in the methods section. Hepatic VEGF expression increased during liver fibrosis development. Neither R-1mAb nor R-2mAb treatment altered VEGF gene expression during development of fibrosis. Control, immunogloblin G treated mice (800 μg/mouse) (G1); R1, R2, R-1mAb and R-2mAb treated mice (800 μg/mouse) (G2 and G3, respectively); R1+R-2, R-1mAb and R-2mAb combination treated group (G4); Oil, corn oil injected negative control mice. Data are means (SD) (n=5).
Histological findings and fibrosis markers
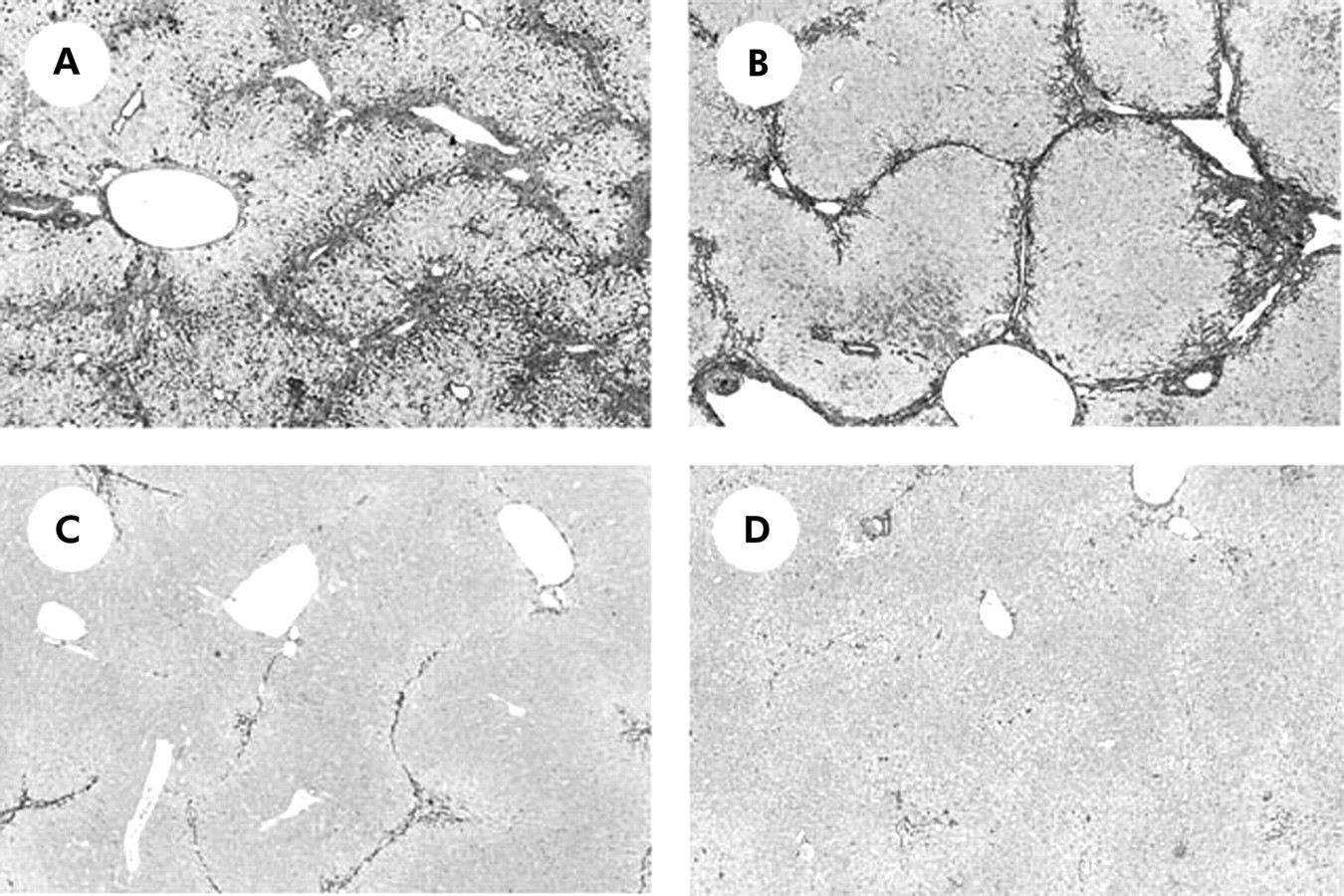
A-M staining revealed that eight weeks of treatment with CCl4 resulted in marked liver fibrosis development (fig 2A). Both R-1mAb and R-2mAb treatment significantly suppressed fibrosis development (fig 2B, 2C, respectively), and the combination treatment of R-1mAb and R-2mAb almost completely attenuated CCl4 induced liver fibrosis (fig 2D). No fibrosis development was found in the corn oil treated group, and the low dose of R-1mAb (400 μg/mouse) did not exert such an inhibitory effect (data not shown). Densitometric analysis showed that the fibrosis areas (fig 3A) mostly corresponded to the histological findings. Although both R-1mAb and R-2mAb significantly suppressed liver fibrosis development compared with the control IgG treated group (p<0.01), the inhibitory impact was more potent with R-2mAb than with R-1mAb treatment (p<0.01). The combination treatment with both mAbs revealed further inhibition compared with that of R-2mAb alone (p<0.05). Hepatic hydroxyproline content showed similar results to those of fibrosis area (fig 3B). These results suggest that both VEGFR-1 and VEGFR-2 play important roles in liver fibrogenesis, and that the role of VEGFR-2 is more predominant than that of VEGFR-1. The serum fibrosis markers, hyaluronic acid and P-III-P, were also significantly suppressed by treatment with R-1mAb and R-2mAb, whereas serum ALT and total bilirubin levels did not change with the use of R-1mAb and R-2mAb (table 1). We also examined the effects of R-1mAb and R-2mAb on the acute liver injury and early liver fibrogenesis step. It has been shown that Masson’s trichrome positive connective tissue accumulation could be observed on day 7 after CCl4 treatment.47 Two or seven days after CCl4 treatment, R-1mAb and R-2mAb did not alter ALT levels in the liver (data not shown). This indicated that the inhibitory effect of mAbs was not a secondary response to a cytoprotective effect against CCl4. Body and liver weights when the mice were killed were not significantly different between the control, R-1mAb treated, and R-2mAb treated groups (data not shown).
Effect of R-1 and R-2 mAb on several markers in animals treated with CCl4

Microphotographs of liver sections from CCl4 treated mice. (A) Control immunogloblin G treated group after CCl4 treatment (800 μg/mouse) (G1). (B, C) R-1mAb and R-2mAb treated (800 μg/ mouse) groups (G2 and G3, respectively). (D) R-1mAb and R-2mAb combination treated group (G4). The livers in G1 show extensive fibrosis development. In G2 and G3, liver fibrosis development was significantly attenuated, and the inhibitory impact was more potent with R-2mAb treatment than with R-1mAb treatment. Fibrosis development was almost completely abolished in the livers of G4 (A-M staining, 40×).

Effects of R-1mAb and R-2mAb on fibrosis area (A) and hepatic hydroxyproline content (B) in the CCl4 treated liver. (A) Fibrosis area was evaluated by an image analyser, as described in the methods section. R-1mAb and R-2mAb significantly suppressed liver fibrosis development compared with the control group (p<0.01), and the inhibitory impact was more potent with R-2mAb treatment than that with R-1mAb treatment (p<0.01). The combination treatment with both mAbs revealed further inhibition compared with that of R-2mAb alone (p<0.05). (B) The inhibitory effects of R-1mAb and R-2mAb on hepatic hydroxyproline content exerted behaviours similar to those on fibrosis area. Control, immunogloblin G treated mice (800 μg/mouse) (G1); R1, R2, R-1mAb and R-2mAb treated mice (800 μg/mouse) (G2 and G3, respectively); R1+R2, R-1mAb and R-2mAb combination treated group (G4); Oil, corn oil injected negative control mice. Data are means (SD) (n=5). *p<0.05, **p<0.01 between the indicated groups.
Neovascularisation
To examine whether the inhibitory effects of R-1mAb and R-2mAb were associated with suppression of neovascularisation in the liver, we evaluated the angiogenic response during liver fibrosis development. We performed a preliminary immunohistochemical analysis of the von Willebrand factor (vWF) related antigen on sections from all experimental groups, and found that R-1mAb/R-2mAb treatment significantly suppressed vWF positive vessels. However, it was hard to accurately evaluate vWF positive cells because of difficulties in identifying the little slit vessels in the R-1mAb and R-2mAb combination treated group (data not shown). It has been reported that CD34 is a more sensitive marker than vWF related antigen.4 CD31 was also shown to be a sensitive marker in EC.48 Among these markers, it has been reported that CD34 expression may be decreased by VEGF.49 We thus used CD31 expression in the current study.
We performed real time PCR analysis of CD31 gene expression to evaluate neovascularisation in the liver. Figure 4 demonstrates that CD31 gene expression was significantly increased in association with liver fibrosis development. Similar to fibrosis area, both R-1mAb and R-2mAb significantly suppressed CD31 gene expression compared with the control group (p<0.01). The inhibitory impact was more potent with R-2mAb treatment than that with R-1mAb treatment (p<0.01), and the combination treatment of both mAbs almost abolished neovascularisation in the liver. Noteworthy was the finding that suppression of angiogenesis by treatment with R-1mAb and R-2mAb was of a similar magnitude to that of inhibition of fibrosis areas.

Effects of R-1mAb and R-2mAb on neovascularisation in the liver. CD31 mRNA expression was examined by real time polymerase chain reaction, as described in the methods section. CD31 gene expression was significantly increased during liver fibrosis development. Treatment with R-1mAb and R-2mAb significantly attenuated neovascularisation in the liver. Suppression of angiogenesis by treatment with R-1mAb and R-2mAb was of a similar magnitude to that of inhibition of fibrosis areas. Control, immunogloblin G treated mice (800 μg/mouse) (G1); R1, R2, R-1mAb and R-2mAb treated mice (800 μg/mouse) (G2 and G3, respectively); R1+R2, R-1mAb and R-2mAb combination treated group (G4); Oil, corn oil injected negative control mice. Data are means (SD) (n=5). *p<0.05, **p<0.01 between the indicated groups.
Effects of R-1mAb and R-2mAb on HSC activation
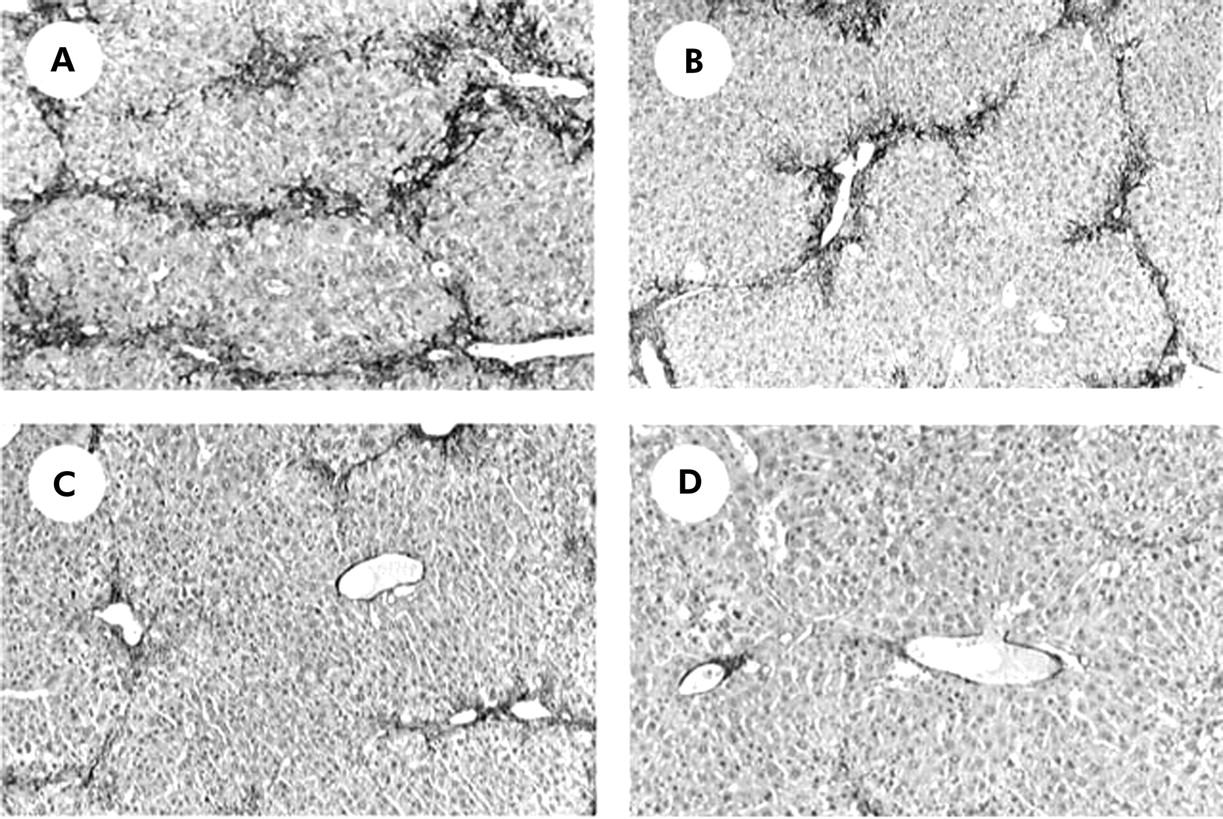
It has been reported that not only SEC, but also activated HSC, express both VEGFR-1 and VEGFR-2.23,24 Immunohistochemical examination showed that α-SMA positive cells were drastically reduced by treatment with R-1mAb and R-2mAb (fig 5A–5D). Computer assisted semiquantitative analysis showed that α-SMA positive cells in the R-1mAb and R-2mAb treated groups were significantly reduced compared with the control group (p<0.01) (fig 6A). We also performed real time PCR analysis to elucidate the effect of these mAbs on α1-(I)-procollagen mRNA expression. R-1mAb and R-2mAb also markedly suppressed mRNA expression of α1-(I)-procollagen in the liver compared with the control group (p<0.01) (fig 6B). The inhibitory effects of R-2mAb on both α-SMA positive cells and α1-(I)-procollagen mRNA expression were significantly stronger than those of R-1mAb (p<0.01). The inhibitory effects of R-1mAb and R-2mAb on α-SMA, α1-(I)-procollagen mRNA expression, and fibrosis area were almost identical, suggesting that suppression of HSC activation also contributed to the antifibrotic effect of these mAbs.

Immunohistochemical analysis of α smooth muscle actin (α-SMA). Immunopositive cells of α-SMA were significantly reduced in the livers of R-1mAb (B), R-2mAb (C), and the combination of R-1mAb and R-2mAb treated groups (D) compared with the control group (A) (G1) (magnification ×40).
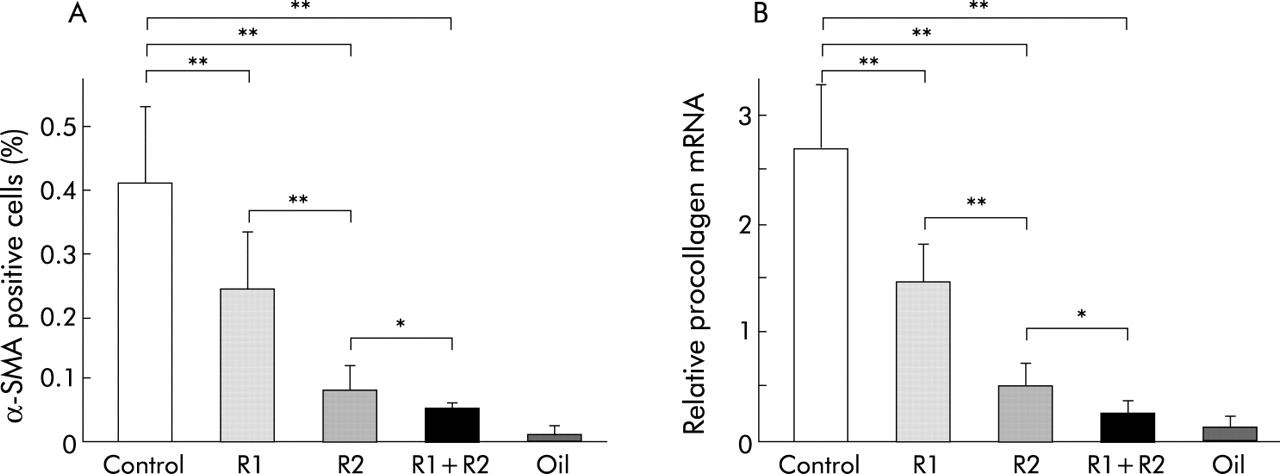
Densitometric analysis of α smooth muscle actin (α-SMA) positive cells (A) and α1-(I)-procollagen mRNA expression (B) in the CCl4 treated liver. α-SMA positive activated hepatic stellate cells and α1-(I)-procollagen mRNA were significantly reduced by R-1mAb and R-2mAb treatment. The inhibitory effect of R-2mAb was more potent than that of R-1mAb. The inhibitory effects of R-1mAb and R-2mAb on α-SMA and α1-(I)-procollagen expression exerted almost parallel reductions. Control, immunogloblin G treated mice (800 μg/mouse) (G1); R1, R2, R-1mAb and R-2mAb treated mice (800 μg/mouse) (G2 and G3, respectively); R1+R2, R-1mAb and R-2mAb combination treated group (G4); Oil, corn oil injected negative control mice. Data are means (SD) (n=5). *p<0.05, **p<0.01 between the indicated groups.
VEGFR-1 and VEGFR-2 receptor activation in situ
To determine whether R-1mAb and R-2mAb at the dose used in the current study (800 μg/mouse) inhibited autophosphorylation in the liver, we investigated tyrosine phosphorylated VEGFR-1 and VEGFR-2 in the liver after intraperitoneally injection of R-1mAb and R-2mAb. R-1mAb and R-2mAb significantly inhibited tyrosine phosphorylation of the respective receptors, and the combination treatment of R-1mAb and R-2mAb almost completely abolished phosphorylation of both receptors in the liver.
Neither activation of VEGFR-1 nor that of VEGFR-2 was altered by administration of R-2mAb and R-1mAb, respectively (fig 7).

Effects of R-1mAb and R-2mAb on the activation of vascular endothelial growth factor (VEGF) receptors VEGFR-1 (fms-like tyrosine kinase (Flt-1)) and VEGFR-2 (kinase-insert domain-containing receptor/fetal liver kinase-1 (Flk-1)). Fifteen minutes after injection of R-1mAb and R-2mAb, the liver was resected from three mice and pooled. The liver lysate was concentrated and used for immunoprecipitation, as described in the methods section. R-1mAb and R-2mAb significantly inhibited tyrosine phosphorylation of the respective receptors. Neither activation of VEGFR-1 nor that of VEGFR-2 was altered by administration of R-2mAb and R-1mAb, respectively. The activation level of VEGFR-1 was lower than that of VEGFR-2. Lane 1, immunogloblin G treated control group (G1); lane 2, R-1mAb treated group (G2); lane 3, R-2mAb treated group (G3); lane 4, R-1mAb and R-2mAb combination treated group (G4); and lane 5, corn oil treated negative control group.
Effect of VEGF on cultured activated HSC and SEC
It has been reported that HSC plated on collagen I are activated progressively whereas those on a basement membrane substratum resembling the normal subendothelial matrix of the liver (EHS matrix) remain quiescent.24,50 We examined the effect of VEGF on HSC proliferation under different culture conditions. Figure 8A shows that both 10 and 100 ng/ml VEGF treatment did not increase in vitro proliferation on an EHS matrix whereas it was stimulated significantly on a collagen I coated dish (p<0.05), indicating that VEGF stimulated activated HSC proliferation but not quiescent HSC. We also examined whether VEGF increased synthesis of the extracellular matrix (ECM) component in activated HSC. As shown in fig 8B, VEGF treatment at a dose of 10 ng/ml significantly upregulated α1-(I)-procollagen mRNA synthesis in activated HSC.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of vascular endothelial growth factor (VEGF) on proliferation and α1-(I)-procollagen mRNA expression of activated hepatic stellate cells (HSC) and hepatic sinusoidal endothelial cells (SEC) in vitro. Cell proliferation and mRNA expression were measured by the MTT assay and real time polymerase chain reaction, as described in the methods section, respectively. (A) At doses of 10 and 100 ng/ml, VEGF treatment did not increase the in vitro proliferation of HSC on an EHS matrix whereas it significantly stimulated proliferation on collagen I (Col-I). *p<0.05 compared with the control group. Control, untreated control group; VEGF, VEGF treated groups at doses of 10 and 100 ng/ml. (B) At a dose of 10 ng/ml, VEGF significantly increased α1-(I)-procollagen mRNA synthesis in activated HSC. **p<0.01 compared with the VEGF untreated group. (C) Proliferation of SEC was significantly increased over time on stimulation with VEGF (10 ng/ml). OD, optical density. *p<0.05, **p<0.01 compared with day 1. Data are means (SD) (n=5).
We next examined the effect of VEGF on SEC proliferation. As it has been shown that VEGF is a survival factor for SEC,45 we chronologically examined the proliferation assay of SEC in the presence of VEGF (10 ng/ml). Figure 8C reveals that proliferation of SEC was significantly increased on stimulation with VEGF over time. Without VEGF, these cells rapidly atrophied and died in a few days. We also examined whether VEGF induced production of factors by SEC that may have an impact on HSC biology, such as platelet derived growth factor and transforming growth factor β. We found that neither factor was increased by treatment with VEGF in SEC (data not shown). In addition to the MTT assay, we performed the [H]3 incorporation experiment for in vitro proliferation of both HSC and SEC. Our results were similar to those of previously reported51 MTT and [H]3 incorporation assays (data not shown).
DISCUSSION
Angiogenesis and fibrosis are key components in development, growth, wound healing, and regeneration. Recent studies have revealed that these processes commonly occur together in many disease states where neovascularisation is believed to initiate the pathological cascade.6 Among the identified angiogenic factors to date, VEGF is one of the most potent and central factors in many physiological and pathological processes.14–16 In liver fibrosis, it has been shown that VEGF expression increased in both human chronic liver diseases and experimental fibrogenesis.10–12 It has also been reported that VEGF expression correlates with chronic liver disease associated angiogenesis and sinusoidal capillarisation.12,52,53 We also observed that VEGF gene expression significantly increased during fibrosis development associated with neovascularisation in the liver, and that suppression of VEGF-receptor interaction significantly attenuated progression of liver fibrosis and angiogenesis.
The biological activities of VEGF are mediated mainly via two type III tyrosine kinase receptors—namely, VEGFR-1 and VEGFR-2—which serve different roles in angiogenesis and signal transduction pathways.15,16,26,28 It has been reported that VEGFR-2 plays a more important role both in vitro and in vivo in several biological events.15,16,35,36 Overexpression of VEGFR-2 in porcine EC caused actin reorganisation, chemotaxis, and mitogenesis in response to VEGF, although VEGFR-1 expression in the same cells had a minimal effect in vitro.28 However, recent studies have revealed that VEGFR-1 is also involved in pathological angiogenesis, such as tumour growth.30–34 In the present study, we found that inhibition of either VEGFR-1 or VEGFR-2 significantly attenuated liver fibrogenesis accompanied by angiogenesis suppression, and that treatment with R-2mAb was more potent than that with R-1mAb. The combination treatment with both mAbs almost completely attenuated liver fibrogenesis. These results indicate that VEGF-receptor interaction is a major regulator of the process of liver fibrosis. Both VEGFR-1 and VEGFR-2 were involved in liver fibrogenesis, and signalling through VEGFR-2 was a predominant pathway compared with that via VEGFR-1.
It is now recognised that activated HSC play an important role in liver fibrosis development.1,54 In addition to EC, recent studies have shown that expression of VEGF and its receptor occurs in activated HSC.22–24 This indicates that the cellular targets of VEGF are not confined to EC, and that VEGF responses reflect the combined effects on both EC and HSC. Recently, it has been shown that hypoxia induced VEGF expression was associated with angiogenesis and liver fibrogenesis. The authors showed that hepatic VEGFR-1 expression increased in liver fibrogenesis, which probably originated from activated HSC.11 The in vitro study showed that VEGFR-1 was selectively increased in activated HSC under hypoxic conditions whereas expression of VEGFR-2 was not upregulated.23 However, it should be noted that VEGFR-2 was constitutively expressed in activated HSC, and that the expression level of VEGFR-2 was much higher than that of VEGFR-1.11,23,24 We found that activated α-SMA positive HSC and α1-(I)-procollagen mRNA were significantly suppressed by R-1mAb and R-2mAb. It has been reported that α-SMA expression in activated HSC may be downregulated by VEGF.21 The authors also claimed that VEGF did not affect proliferation of activated HSC in vitro. In contrast, we and others have shown different results under different culture conditions,24 suggesting that VEGF exerts a different biological effect on activated HSC under different culture conditions. After liver injury, HSC proliferate and become activated to the myofibroblastic phenotype, which is characterised by an increase in expression of α-SMA.1 As it is now widely recognised that α-SMA is a useful and reliable marker for in vivo fibrogenesis,1,38,40 we also employed this marker in the current study.
We also found that VEGF stimulated proliferation of activated HSC in vitro, indicating that VEGF-receptor interaction in HSC also plays an important role in liver fibrogenesis. We found that HSC did not respond to VEGF when they were cultured on Matrigel. Matrigel may sequester VEGF as it can bind with the proteoglycans of Matrigel. However, we found similar effects with low and high exogenous VEGF treatment (10 ng/ml and 100 ng/ml, respectively). With high doses of VEGF, we assume that VEGF can act on HSC even if Matrigel may sequester some part of VEGF. A similar effect of VEGF on activated HSC in Matrigel has been reported.24
It would be important to elucidate the binding sites of R-1mAb and R-2mAb during liver fibrogenesis. We attempted to localise these binding sites of mAbs by immunohistochemical double staining but we failed to obtain good results. The background was very intense, and interpretation was very difficult (data not shown). Furthermore, the specific role of each receptor in activated HSC remains obscure at this time as we could not obtain specific R-1mAb and R-2mAb for the rat. When mAbs against rat VEGFR-1 and VEGFR-2 become available, further studies may elucidate the role of each receptor in liver fibrogenesis.
VEGF was originally identified as a vascular permeability factor that increased microvessel permeability approximately 50 000 times more than histamine.14,16,27 It induces extravasation of plasma proteins, leading to an increase in the ECM. It has also been reported that VEGF increased mRNA levels of connective tissue growth factor, which has known actions on ECM production.55 In the current study, we found that VEGF significantly increased α1(I)-procollagen mRNA in activated HSC, indicating that VEGF stimulates liver fibrogenesis through both ECM production and proliferation in activated HSC. Nevertheless, it is well known that HSC are the primary ECM producing cell type but SEC also respond rapidly to injury by synthesising an isoform of the cellular fibronectin that stimulates HSC activation.56 They also produce ECM components, such as type IV collagen and several proteoglycans.57,58 In addition to its angiogenic activity, VEGF may also stimulate fibrosis development through these biological activities.
In summary, hepatic VEGF mRNA expression was significantly increased during the development of fibrosis, and both R-1mAb and R-2mAb treatment significantly attenuated the fibrogenesis associated with suppression of neovascularisation in the liver. The inhibitory effect of R-2mAb was more potent than that of R-1mAb, and the combination treatment with R-1mAb and R-2mAb almost completely attenuated liver fibrosis development. α-SMA positive activated HSC and α1-(I)-procollagen mRNA were significantly suppressed by R-1mAb and R-2mAb, and VEGF stimulated proliferation of activated HSC in vitro. These results suggest that interaction between VEGF and its receptors, which reflected the combined effects of both on HSC and SEC, was a prerequisite for liver fibrosis development.
Acknowledgments
This work was supported in part by a Grant-in-Aid for Scientific Research (C-14570498) from the Japanese Ministry of Education, Science, Sports, and Culture.