Article Text

Abstract
Objective The authors tested whether the anti-interleukin (IL)-17A monoclonal antibody secukinumab was safe and effective for the treatment of active Crohn's disease.
Design In a double-blind, randomised, placebo-controlled proof-of-concept study, 59 patients with moderate to severe Crohn's disease (Crohn's Disease Activity Index (CDAI) ≥220 to ≤450) were assigned in a 2:1 ratio to 2×10 mg/kg intravenous secukinumab or placebo. The primary end point, addressed by Bayesian statistics augmented with historical placebo information, was the probability that secukinumab reduces the CDAI by ≥50 points more than placebo at week 6. Ancillary analyses explored associations of 35 candidate genetic polymorphisms and faecal calprotectin response.
Results 59 patients (39 secukinumab, 20 placebo, mean baseline CDAI 307 and 301, respectively) were recruited. 18/59 (31%) patients discontinued prematurely (12/39 (31%) secukinumab, 6/20 (30%) placebo), 10/59 (17%) due to insufficient therapeutic effect (8/39 (21%) secukinumab, 2/20 (10%) placebo). Fourteen serious adverse events occurred in 10 patients (seven secukinumab, three placebo); 20 infections, including four local fungal infections, were seen on secukinumab versus none on placebo. Primary end point analysis estimated <0.1% probability (∆CDAI (SD) =33.9 (19.7), 95% credible interval −4.9 to 72.9) that secukinumab reduces CDAI by ≥50 points more than placebo. Secondary area under the curve analysis (weeks 4–10) showed a significant difference (mean ΔCDAI=49; 95% CI (2 to 96), p=0.043) in favour of placebo. Post hoc subgroup analysis showed that unfavourable responses on secukinumab were driven by patients with elevated inflammatory markers (CRP≥10 mg/l and/or faecal calprotectin≥200 ng/ml; mean ΔCDAI=62; 95% CI (−1 to 125), p=0.054 in favour of placebo). Absence of the minor allele of tumour necrosis factor-like ligand 1A was strongly associated with lack of response measured by baseline-adjusted changes in calprotectin at week 6 (p=0.00035 Bonferroni-corrected).
Conclusions Blockade of IL-17A was ineffective and higher rates of adverse events were noted compared with placebo.
Clinical trial registration This trial was registered at ClinicalTrial.gov with the number NCT01009281.
- Crohn's disease
- clinical trials
- inflammatory bowel disease
- ulcerative colitis
- genetic polymorphisms
- genetics
- genotype
- pharmacogenetics
- statistics
- chronic IBD
- antibody targeted therapy
- immune response
- antibacterial mucosal immunity
- mucosal defense
- antibacterial peptide
- IBD basic research
- gut immunology
- gut inflammation
- cytokines
- immunoregulation
- immunology
- inflammation
- inflammatory mediators
- pharmacokinetics
- pharmacology
- infliximab
- 5-aminosalicylic acid
Statistics from Altmetric.com
- Crohn's disease
- clinical trials
- inflammatory bowel disease
- ulcerative colitis
- genetic polymorphisms
- genetics
- genotype
- pharmacogenetics
- statistics
- chronic IBD
- antibody targeted therapy
- immune response
- antibacterial mucosal immunity
- mucosal defense
- antibacterial peptide
- IBD basic research
- gut immunology
- gut inflammation
- cytokines
- immunoregulation
- immunology
- inflammation
- inflammatory mediators
- pharmacokinetics
- pharmacology
- infliximab
- 5-aminosalicylic acid
Significance of this study
What is already known on this subject?
-
Conventional treatment of Crohn's disease includes immunosuppression with corticosteroids, thiopurines or methotrexate, and antitumour necrosis factor therapy for patients with persistent disease activity.
-
Data obtained in animal models of inflammatory bowel disease suggest involvement of interleukin (IL)-17 in Crohn's disease pathogenesis, and overexpression of IL-17 was observed in intestinal tissue from patients with active Crohn's disease.
-
Genetic studies found a strong association of IL-23R with susceptibility to Crohn's disease, thus implicating the IL-23–IL-17 axis in disease pathogenesis.
-
Secukinumab (AIN457), a fully human selective anti-IL-17A monoclonal antibody, has completed phase II trials in psoriasis and rheumatoid arthritis and was found to be safe and efficacious in these patient populations.
What are the new findings?
-
Blockade of interleukin-17A was ineffective and higher rates of adverse events were noted compared with placebo in this proof-of-concept study.
-
In an exploratory substudy, lack of response as measured by faecal calprotectin was associated with a polymorphism in the tumour necrosis factor-like ligand gene.
How might it impact on clinical practice in the foreseeable future?
-
Results from this small proof-of-concept study suggest that no benefit is to be expected from blockade of interleukin-17A in patients with moderate-to-severe inflammatory Crohn's disease.
Introduction
Conventional treatment of Crohn's disease includes immunosuppression with corticosteroids, thiopurines or methotrexate (MTX), and antitumour necrosis factor therapy (ATT) for patients with persistent disease activity.1 Although most patients with moderate-to-severe Crohn's disease respond to ATT, secondary failures due to intolerance or loss of initial response are common.2–4 Thus, additional therapies with new mode of mechanisms are required.5
Studies in animal models identified a pivotal role of interleukin-23 (IL-23) in promoting intestinal inflammation via inflammatory mediators including IL-6 and IL-17A.6–8 Consistent with a more complex immunoregulatory role of IL-17A in inflammatory bowel disease (IBD), results obtained from other animal models also suggested protective functions of IL-17A in IBD.9–11 In Crohn's disease, increased expression of IL-17A mRNA and intracellular protein in the intestinal mucosa has been reported.12 Elevated faecal IL-17A levels were described in active Crohn's disease, together with increased numbers of IL-23 and IL-17A producing cells within the lamina propria of Crohn's patients.13 In 2006, The North American IBD Genetics Consortium was the first to report in a genome-wide association study a strong genetic association of IL-23R with susceptibility to Crohn's disease.14 Further evidence of this association was subsequently seen in independent large genome-wide association study cohorts,15 ,16 implicating the IL-23–IL-17 axis in disease pathogenesis and providing indirect evidence for a role of IL-17A in Crohn's disease.
Phase II trials in Crohn's disease have reported that blockade of p40, the shared subunit of the T helper cell 1 (Th1) cytokine IL-12 and the Th17 modulator IL-23, is effective.17 ,18 Although anti-p40 antibodies were assumed to abrogate interferon γ mediated intestinal inflammation through blockade of IL-12 it is now thought that inflammation attributed to IL-12 is mediated by IL-23 and its downstream cytokines IL-17A and IL-22. Therefore, inhibition of IL-17A represents a potential approach for treating active Crohn's disease. Consequently, a proof-of-concept study in Crohn's disease was initiated with secukinumab (AIN457), a fully human selective anti-IL-17A monoclonal antibody that has completed phase I–II trials in psoriasis and rheumatoid arthritis (RA).19
Methods
Patients and study design
A global, multicentre, randomised, double-blind, placebo-controlled parallel-group phase IIa trial was conducted at 25 centres in Europe (Poland, Germany and Austria), the USA and Canada. Institutional review boards or ethics committees at participating centres approved the protocol before the study was started. All patients provided written informed consent. A separate written consent was required for DNA sampling and analysis. Men and women aged 18–75 with moderate to severe Crohn's disease (Crohn's Disease Activity Index, CDAI score ≥220 to ≤450) were eligible. Key inclusion criteria included diagnosis of Crohn's disease20 ≥3 months before screening. Objective evidence of inflammation at baseline was defined by the presence of C reactive protein (CRP) ≥10 mg/l and/or faecal calprotectin (FCP) ≥200 ng/ml. Patients with any CRP or FCP value were eligible. Concurrent therapies with stable doses of azathioprine, mercaptopurine, MTX or steroids (prednisolone ≤40 mg/dl or equivalent) were permitted. Tapering of immunosuppressants was not allowed. Patients with stricture causing obstructive symptoms, or fistulising disease complicated by infection, a positive (>5 mm) tuberculin skin test or biologics therapies within the preceding 2 months (except 3 months for certolizumab) were excluded.
After a screening period of 28 days, patients were assigned, in a ratio of 2:1, to receive either secukinumab given as a dose of 10 mg/kg or matching placebo administered as 2 h intravenous infusions on days 1 and 22. Safety and efficacy assessments were done at weeks 1, 2, 3, 4, 5, 6, 10, 14 and 18 (or end of study). The primary end point was the effect of secukinumab on mean CDAI scores at week 6. Secondary end points included efficacy at weeks 2 and 4, area under the response curve (AUC) between weeks 4 and 10, whereby week 4 was chosen as the predicted time-point of maximum concentration, C(max), and percentage of patients achieving remission (defined as CDAI <150) or achieving response defined as ≥100 points reduction in CDAI scores. Due to poor recruitment, several protocol amendments were implemented. The study was expanded into Europe; patients on concomitant MTX, azathioprine or 6-mercaptopurine with doses kept stable for at least 10 weeks prior to enrolment and patients with normal CRP levels, who had been excluded in the first version of the protocol, were allowed to participate.
Thirty-five genetic polymorphisms were selected to be tested for association with response to secukinumab. Of these, the vast majority was selected because of compelling statistical evidence of association with Crohn's disease16 ,21 (supplementary file 3), the underlying hypothesis being that single nucleotide polymorphisms (SNPs) associated with disease risk may identify different disease subtypes that may lead to differential response to therapy. SNPs in the IL-17A gene region were selected due to IL-17A being the principle target of secukinumab. All genotyping was performed by Novartis, SNP genotyping using TaqMan, and human leucocyte antigen genotyping using a combination of sequence based typing, sequence specific oligonucleotide and sequence specific primers. All patients who signed a consent form for this substudy and provided a DNA sample were eligible for inclusion.
Randomisation and masking
Randomisation was by validated Interactive Voice Response System that automated the random assignment of patient numbers to randomisation numbers and was kept strictly confidential. Randomisation was stratified by baseline CRP level (normal or elevated) once patients with normal CRP were eligible.
Statistical methods
The planned sample size of 60 patients, 40 on secukinumab and 20 on placebo, was determined by simulation studies.
The primary analysis was based on a Bayesian analysis estimating the CDAI reduction at week 6 under secukinumab, placebo and their difference. Bayesian analysis results are reported as probabilities with credible intervals, in contrast to frequentist analyses that report p values and CIs.22 The primary analysis was augmented by historical information of effects of placebo treatment from six Crohn's disease studies.23–28 To account for between-trial variation, the historical information from the 671 placebo patients in these studies was down weighted to a strength of information corresponding to 20 patients.29 For further details on Bayesian analysis and sample size, please see the study protocol (supplementary file 1).
Two scheduled interim analyses (IAs) were performed when 17 and 41 patients, respectively, had passed their 6-week time-point. The purpose of the second IA was to allow study termination if criteria for success or futility had been met.
A prespecified criterion for futility was defined as ≥90% probability that secukinumab reduces the CDAI by <40 points more than placebo. Results were defined as positive ‘proof-of-concept’ if at the planned interim or final analysis the Bayesian model estimated ≥95% probability that secukinumab reduces the CDAI more than placebo and ≥50% probability that secukinumab reduces the CDAI by ≥50 points more than placebo.
All secondary analyses were not Bayesian in nature and did not include historical placebo information. Secondary analyses included comparisons between the treatment arms of: (a) AUC between weeks 4 and 10 (analysis of covariance with baseline CDAI as covariate); (b) remission rates, with remission defined as a CDAI of ≤150 (χ2 test); and (c) response rates, with response defined as a CDAI reduction from baseline by ≥100 points (χ2 test).
Subgroup analysis of patients (post hoc) with or without markers of inflammation was performed for AUC weeks 4–10, as described above.
An exploratory analysis of genetic data was performed on the subgroup of patients who consented to participate in this substudy. The 35 polymorphisms were tested individually for association with change in FCP from baseline to week 6. One placebo-treated patient was excluded due to an extreme outlying value. All statistical tests were performed using linear regression models, with change from baseline to week 6 in FCP as the dependent variable, the number of copies of the minor (ie, less common) allele carried by the individual as the predictor and baseline FCP as a covariate. Other potential covariates were evaluated but excluded due to lack of evidence of association with change in FCP. All statistical tests were two-tailed, with no imputation for missing data. Because of the small sample size, a permutation test was performed to confirm the accuracy of the most significant p value. In this test, genotype was randomly permuted 100 000 times, and the regression model described above was applied to each permutation. The percentage of permutations yielding a p value at least as significant as that obtained from the actual data was taken as the empirical p value. p Values reported in results and shown in figure 3 were Bonferroni-corrected for multiple comparisons.
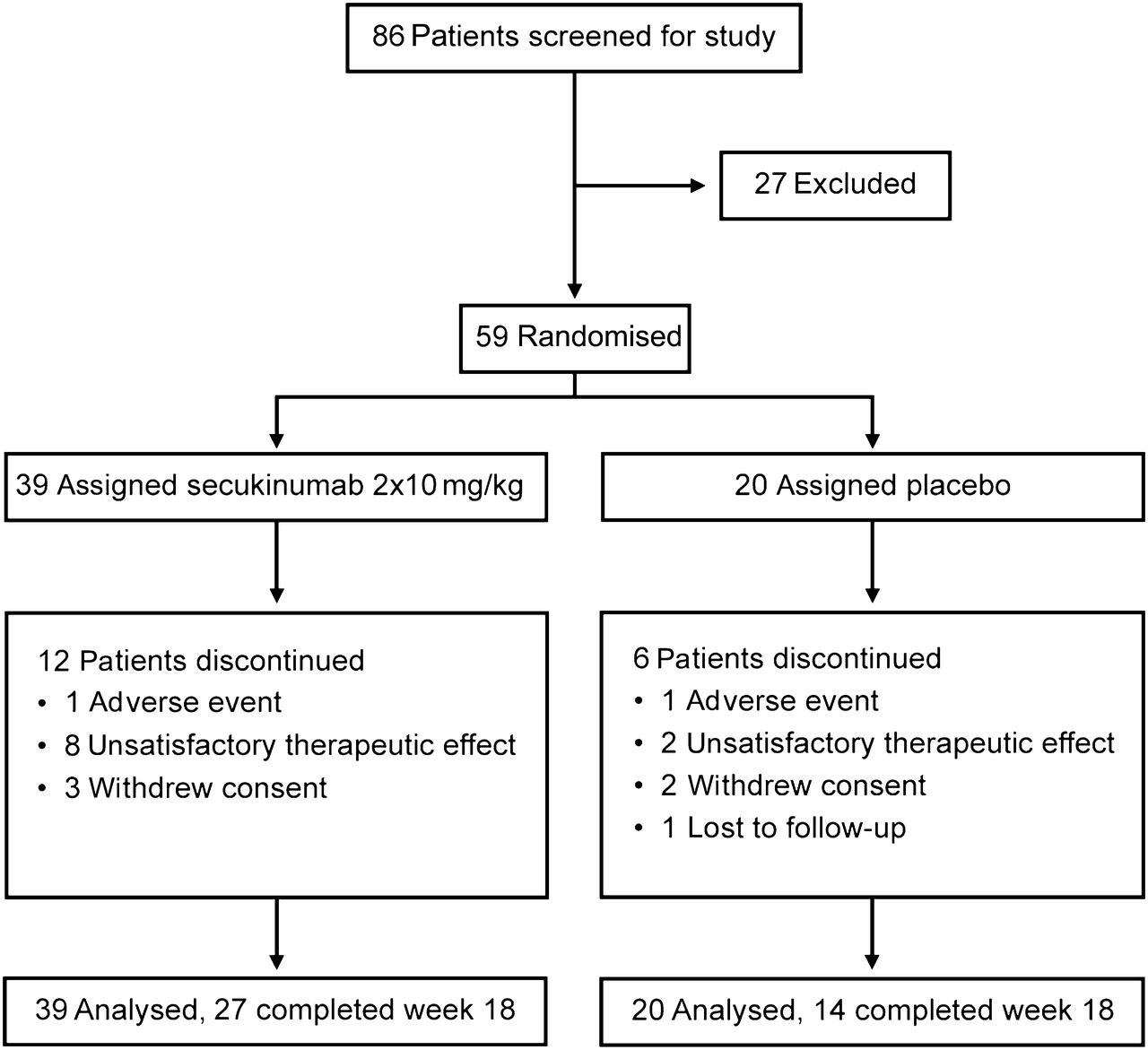
Trial profile.
FCP ELISA: Matrix extracts were prepared from human faeces samples by homogenisation. A validated in-house calprotectin ELISA was applied to determine concentrations of FCP.
Results
Figure 1 shows the study flow chart of 59 patients enrolled between August 2008 and May 2010 and randomly allocated to secukinumab (n=39) or placebo (20); 18 terminated study participation early and three discontinued study drug before the second infusion. Baseline characteristics were similar between treatment groups (table 1) with the exception of prior ATT and bowel surgery. Disease location was reported for 55/59 patients, whereby five patients had colonic involvement and 10 patients isolated ileal disease. The trial was stopped prematurely after a planned IA of 41 patients because the prespecified criterion for futility was met. A further 18 patients were enrolled before completion of the IA, and hence the final analysis was based on data from 59 patients. All patients were included in the intention-to-treat analysis.

Mean (±SE) Crohn's Disease Activity Index score over time.
Baseline characteristics
Primary end point analysis at week 6 using Bayesian analysis concluded that the probability of secukinumab being superior to placebo was 4.4% (table 2).
Primary end point (Bayesian analysis including historical controls)
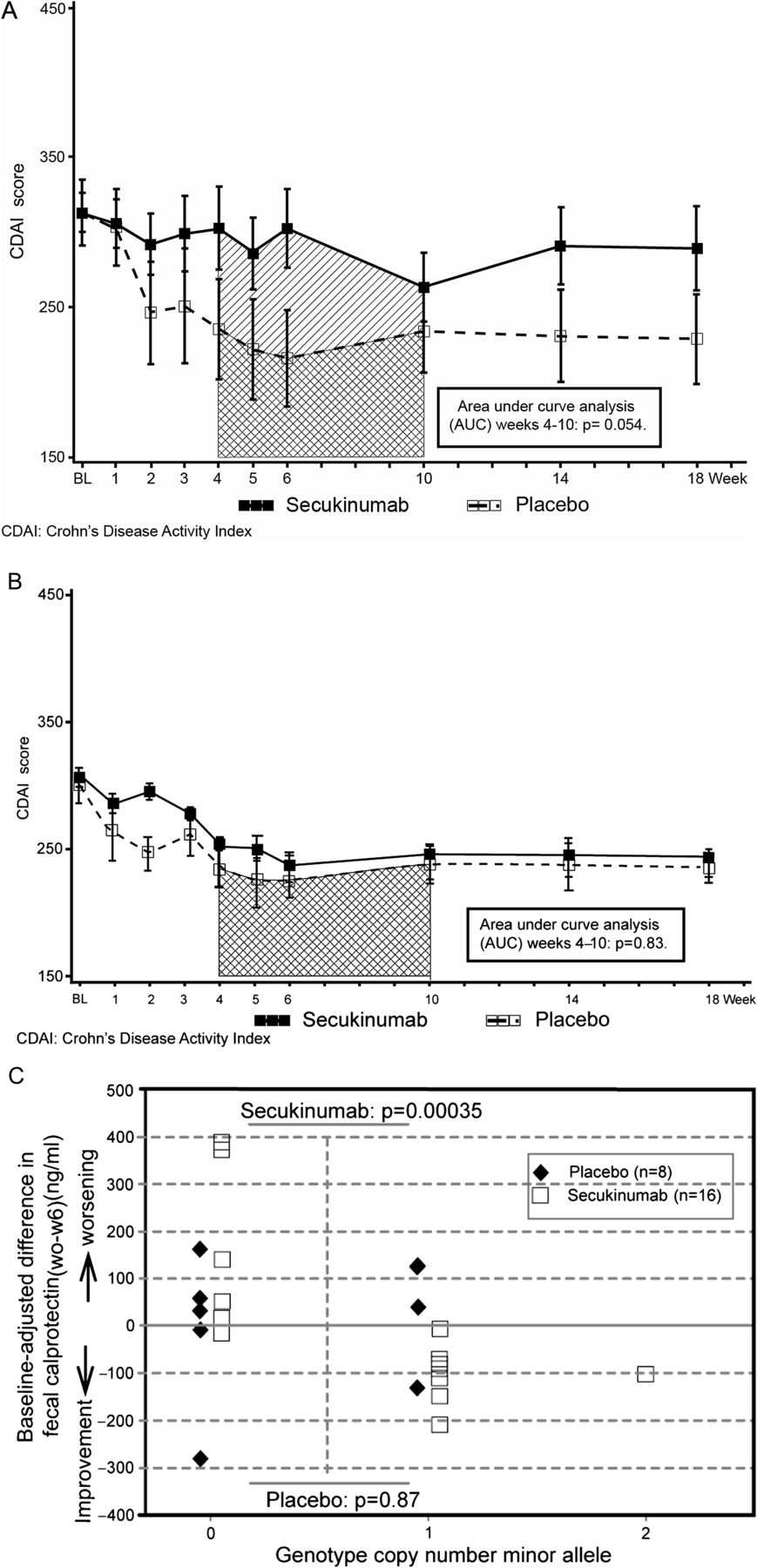
The difference in mean CDAI change from baseline between secukinumab and placebo was 33.9 points (SD 19.7, 95% credible interval −4.9 to 72.9), as reduction in the CDAI in the placebo group (−63.1 points) was greater than in the secukinumab group (−29.2 points, figure 2).

{kind=link}
{kind=link}
{kind=link}
(A). Mean (±SE) Crohn's Disease Activity Index score over time, patients with elevated markers of inflammation. (B). Mean (±SE) Crohn's Disease Activity Index score over time, patients without elevated markers of inflammation. (C). Exploratory pharmacogenetic analysis.
None of the comparisons at secondary time points demonstrated significant differences (supplementary file 2), but AUC analysis between weeks 4 and 10 showed a statistically significant difference of mean CDAI change between secukinumab and placebo in favour of placebo (p=0.043, figure 2).
To explore the apparent adverse effect of secukinumab on Crohn's disease activity, post hoc analysis compared response to secukinumab or placebo in patients with and without objective evidence of inflammation defined by CRP or FCP. A strong trend in favour of placebo-treated patients was seen for comparisons of secukinumab and placebo within the inflammatory subgroup (n=33; mean ΔCDAI =62 (95% CI −1 to 125, p =0.054, figure 3A) but not within the non-inflammatory subgroup (n=26; p=0.87, figure 3B), indicating that the unfavourable response on secukinumab in the entire cohort was overall driven by patients with inflammatory Crohn's disease.
Exploratory analysis of 35 Crohn's disease candidate genetic polymorphisms (supplementary file 3) was performed in a subgroup of 24 patients (16 secukinumab, eight placebo). A polymorphism (rs4263839) in an intron of the tumour necrosis factor-like ligand 1A (TL1A) gene demonstrated a highly significant association with FCP response among secukinumab-treated patients (p=0.00035 in permutation test after Bonferroni correction for multiple comparisons; figure 3, panel C), but not in placebo-treated patients (p=0.87). Absence of the minor allele was associated with worsening (ie, increase in FCP concentrations).
Table 3 summarises safety data up to week 18 (end of study). More patients experienced any adverse event in the secukinumab than in the placebo groups: 29/39 (74%) and 10/20 (50%) patients, respectively, especially for infections: Twenty infections were seen in 17/39 (44%) patients on secukinumab, including four local fungal infections in four patients, versus none on placebo. Severe adverse event were reported in 11/39 (28%) in the secukinumab and 2/20 (10%) in the placebo groups. Fourteen serious adverse events (SAEs) occurred in 10 patients; 7/10 were reported as drug-related, six in the secukinumab and one in the placebo groups. Of the seven SAEs suspected to be drug related, five were cases of worsening of Crohn's disease, four on secukinumab and one on placebo. Two additional SAEs (pilonidal cyst, ileostomy) were observed on secukinumab; these SAEs occurred 24 days and 7 days respectively after discontinuation from study, and therefore were not reported in the clinical database (not shown in table 3).
Adverse events (AEs) overall and most frequent events: n (%) of subjects (>5% in any group)
Both SAEs were considered by the investigator to be related to worsening of underlying Crohn's disease. Infections that were suspected by the investigator to be drug related included one case each of candidiasis, fungal infection and aphthous stomatitis. Few laboratory abnormalities were noted on secukinumab, apart from neutropenia in 4/39, compared with 1/20 on placebo, without associated infection. One patient each on secukinumab and placebo had mild abnormalities in a single liver function test.
Discussion
Secukinumab therapy failed to reduce mean CDAI by ≥50 points more than placebo at week 6, missing the primary end point and fulfilling the prespecified criterion for futility in the IA. As a consequence, the study was terminated prematurely. The results also suggest that inhibition of IL-17A may have adverse outcomes in a subgroup of patients with objective evidence of inflammation.
It is possible that blocking of IL-17A may interfere with a protective function of IL-17A in the intestine. Although animal models of IBD and genome-wide association studies suggest an important role of IL-17A in perpetuating chronic inflammation by activated T cells,30 protective roles have also been postulated based on T cell dependent9 and T cell independent models of colitis.11 Results obtained in this study of patients with active Crohn's disease differ from those obtained in phase II studies of secukinumab in RA19 ,31 and psoriasis32 where a beneficial effect of treatment was observed. Moreover, increased rates of adverse events and infections seen in this small study were not seen in these larger studies of secukinumab in RA and psoriasis. Recent reports in patients with immune deficiencies suggested that genetic defects affecting the IL-17 signalling pathway may be associated with occurrence of chronic mucocutaneous candidiasis (CMC),33–35 a condition characterised by persistent or recurrent disease of the nails, skin, oral or genital mucosae caused by candida albicans species.36 Although subjects in these studies had multiple immune defects arising from their mutations, these studies may raise the possibility that treatments targeting IL-17A could be associated with higher rates of CMC. In our current study of patients with Crohn's disease, a higher frequency of fungal infections was indeed seen in patients on secukinumab (9.1%) compared with placebo (0%). In contrast, rates of fungal infections were as expected and comparable between secukinumab and placebo arms in the larger phase IIb studies in psoriasis and RA, and no cases of CMC were reported in either of these two studies (manuscripts submitted and under review). Although the explanation for these discordant results for both efficacy and safety is unknown, it is possible that the immunological environments in RA and psoriasis are distinct from Crohn's disease where the microbiota plays a critical role in disease expression.
Patients treated with secukinumab had persistent disease activity (measured by AUC analysis of the CDAI response) compared with placebo, and subgroup analysis of inflammatory versus non-inflammatory Crohn's disease showed that this was more prevalent in the patients with objective markers of inflammation (CRP or FCP). This is consistent with higher rates of discontinuations due to lack of efficacy and higher rates of adverse events on secukinumab, including SAEs of worsening of Crohn's disease suspected to be study drug related. Unlike our secukinumab results, post hoc analysis of ustekinumab (an anti-IL-12/IL-23 antibody) in Crohn's disease suggests that patients with elevated CRP, colonic disease and prior ATT had better response rates,37 prompting a trial in patients with active Crohn's disease who have failed or developed intolerance to ATT.38 Prior ATT exposure did not contribute to differential outcomes in our study (data not shown). Exploratory analysis showed an association between persistent gut inflammation after secukinumab, as measured by calprotectin, and a polymorphism of TL1A. The TL1A gene encodes a cytokine that drives pathogenic T cells in various autoimmune inflammatory processes and has been implicated in IBD.39 ,40
One limitation of our proof-of-concept study was the small sample size, and post hoc and exploratory subgroup analyses were based on small patient numbers. A second limitation was a potential bias towards more severe disease in the secukinumab group based on higher rates of prior ATT and bowel surgery (table 1). However, observed responses did not differ between patients with or without prior ATT or with or without prior bowel surgery. A third limitation arose from the change in the study population to allow inclusion of patients with CRP levels below 10 mg/l, and background immunosuppressive therapy. To avoid an imbalance between the secukinumab and placebo groups in CRP, a potential prognostic factor that could influence response, a randomisation stratifier based on CRP, was introduced with the study amendment to obtain balanced strata with respect to this potential confounder. It is noteworthy that the futility stopping criterion was met even with this broader population, although the unfavourable response on secukinumab was mainly driven by patients with inflammatory Crohn's disease.
Last, the exploratory genetic analysis was based on a small sample size and it cannot be excluded that findings are spurious. Therefore, these preliminary observations require confirmation in an independent cohort of Crohn's patients before definitive conclusions can be drawn. However, the very low p value suggests that the observed difference may be relevant, and findings may serve as a basis for new hypothesis testing.
Inhibition of IL-17A by secukinumab was ineffective in this trial of patients with moderate to severe Crohn's disease. Compared with placebo, secukinumab had a smaller effect and patients on secukinumab showed higher rates of adverse events. However, this was a small exploratory study, and adverse outcomes observed in this study were not seen in trials of secukinumab in other indications. It may be that IL-17A has a protective function in inflammatory Crohn's disease, possibly more pronounced in patients without the minor TL1A variant (rs4263839A).
Acknowledgments
We thank Michael Shetzline, MD, and Franco di Padova, MD, Novartis Institutes for BioMedical Research, Basel, Switzerland for their contributions to the design of the study; and Amanda Wang, PhD, Novartis Institutes for BioMedical Research, Cambridge, MA, USA, who helped with aspects of the pharmacogenetic analysis.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
Footnotes
-
Funding Novartis Pharma AG, Basel, Switzerland funded this study. External advisors (B Feagan, P D R Higgins, M and J Karczewski, W Reinisch, B Sands, S Travis, J Wehkamp, M Yao) together with the NIBR clinical team supervised the study and assisted with data interpretation. Investigators (see also Acknowledgements) gathered data and Novartis staff entered them into the clinical database. Novartis statisticians and programmers together with Covance statisticians did the analyses, and advisory committee members together with Novartis experts prepared this manuscript. The corresponding author had full access to all data and had final responsibility for the decision to submit for publication.
-
Competing interests Dr Wolfgang Hueber is an employee of Novartis Pharma and owns shares; Dr Bruce E Sands received consulting fees for service on a scientific advisory board for Abbott Immunology, Avaxia Biologics, Bristol-Myers Squibb, Elan Pharmaceuticals, Glaxo SmithKline Welcome, Novartis Pharmaceuticals, Pfizer and Prometheus Laboratories; consulting fees from Emmi Solutions; and holds common stock in Avaxia Biologics (a company that is not publicly traded); Steve Lewitzky is an employee of Novartis Pharma; Dr Marc Vandemeulebroecke is an employee of Novartis Pharma and owns Novartis shares; Dr Walter Reinisch is a medical advisor to Novartis; Dr Peter D R Higgins consults for Amgen, Genentech and JBR Pharma, and receives honoraria from Abbott; Dr Jan Wehkamp has no conflict of interest; Dr Brian G Feagan has been a scientific advisor for Protein Design Labs, Astra Zeneca, Elan/Biogen, Celltech, Synta, Merck, Celgene, Novartis, Given Imaging Inc., UCB Pharma, Salix Pharmaceuticals, Abbott Laboratories, Centocor Inc. Pfizer, Axcan, Tillotts Pharma AG, Prometheus Laboratories, a consultant for Synta, Millennium, Merck, Centocor, Elan/Biogen, Janssen-Ortho, Protein Design Labs, ISIS, Teva Pharmaceuticals, Santarus, Bristol-Myers Squibb, Celgene, UCB Pharma, Abbott, Proctor and Gamble, Genentech, Tillotts, Given Imaging Inc., Salix Pharm., Ore Pharm. (previously GeneLogic), Novo Nordisk, GSK, Actogenix, Prometheus Therapeutics and Diagnostics, Athersys, Alba Therapeutics, Axcan, Pfizer, Shire, Wyeth, Zealand Pharm and has received a research grant from Merck, Milllennium, Tillotts, Abbott, Engelheim, Novartis, Centocor, Synta, Elan/Biogen, UCB Pharma, BMS, Proctor and Gamble, Genentech, CombinatoRx, ActoGeniX; Dr Michael D Yao has nothing to disclose; Dr Marek Karczewski was an external consultant for Novartis Pharma AG; Dr Jacek Karczewski was an external consultant for Novartis Pharma AG; Nicole Pezous is an employee of Novartis Pharma; Dr Stephan Bek is an employee of Novartis Pharma; Dr Gerard Bruin is an employee of Novartis Pharma and owns Novartis shares; Dr Bjoern Mellgard is an employee of Novartis Pharma; Claudia Berger is an employee of Novartis Pharma and owns Novartis shares; Dr Marco Londei is an employee of Novartis Pharma; Dr Arthur P Bertolino is an employee of Novartis Pharma; Dr Gervais Tougas is an employee of Novartis Pharma and owns Novartis shares; Dr Simon P L Travis has received honoraria for advisory boards or consultancy from Abbott; Asahi; Aspreva; BMS; Centocor; Cosmo; Elan; Ferring; Genentech; Genzyme; Giuliani; GSK; Glenmark; MSD; Novartis; Ocera; Procter & Gamble Pharmaceuticals; PDLBiopharma; Santarus; Schering-Plough; Shire; Takeda; Tillotts; UCB Pharma; Vertex; Vifor and Warner Chilcott. He has given expert testimony on behalf of Elan, Cosmo, Ocera, Procter & Gamble, Santarus and Tillotts, and received unrestricted educational grants from Abbott, Ferring, MSD, Procter & Gamble, Schering Plough and Warner Chilcott.
-
Patient consent Obtained.
-
Provenance and peer review Not commissioned; externally peer reviewed.