Article Text

Abstract
Objectives: To study myopathies with serum antibodies to the signal recognition particle (SRP), an unusual, myositis specific antibody associated syndrome that has not been well characterised pathologically.
Methods: Clinical, laboratory, and myopathological features were evaluated in seven consecutive patients with a myopathy and serum anti-SRP antibodies, identified over three years. The anti-SRP myopathy was compared with myopathology in other types of inflammatory and immune myopathies.
Results: The patients with anti-SRP antibodies developed weakness at ages ranging from 32 to 70 years. Onset was seasonal (August to January). Weakness became severe and disability developed rapidly over a period of months. Muscle pain and fatigue were present in some patients. No patient had a dermatomyositis-like rash. Serum creatine kinase was very high (3000 to 25 000 IU/l). Muscle biopsies showed an active myopathy, including muscle fibre necrosis and regeneration. There was prominent endomysial fibrosis, but little or no inflammation. Endomysial capillaries were enlarged, reduced in number, and associated with deposits of the terminal components of complement (C5b-9, membrane attack complex). Strength improved in several patients after corticosteroid treatment.
Conclusions: Myopathies associated with anti-SRP antibodies may produce severe and rapidly progressive weakness and disability. Muscle biopsies show active myopathy with pathological changes in endomysial capillaries but little inflammation. Corticosteroid treatment early in the course of the illness is often followed by improvement in strength. In patients with rapidly progressive myopathies and a high serum creatine kinase but little inflammation on muscle biopsy, measurement of anti-SRP antibodies and pathological examination of muscle, including evaluation of endomysial capillaries, may provide useful information on diagnosis and treatment.
- myositis
- signal recognition particle
- antibodies
Statistics from Altmetric.com
Immune mediated and inflammatory myopathies are usually classified according to their clinical, electrophysiological, serological, and pathological features.1–3 Polymyositis is a group of disorders that have in common weakness (usually proximal), a raised serum creatine kinase, a myopathic and irritative pattern of changes on electrodiagnostic testing, and myopathic changes—often associated with inflammation—on pathological examination of muscle. Dermatomyositis has many features that are similar to polymyositis but is distinguished by skin involvement and by muscle pathology that usually includes perifascicular myopathic changes. Inclusion body myositis is distinguished from other inflammatory myopathies by its clinical pattern of patchy, asymmetrical, proximal and distal weakness and pathological changes that include vacuoles and focal invasion of muscle fibres by inflammatory cells. Polymyositis and dermatomyositis are generally thought to be immune mediated, but the pathogenic processes underlying inclusion body myositis are unclear.
It now appears that subgroups of polymyositis and dermatomyositis can be delineated on the basis of clinical, serological, and pathological features of disease syndromes. Some of these myopathy syndromes are associated with characteristic “myositis specific” serum antibodies.4,5 The best defined, and most common, of these syndromes is associated with serum antibodies to the Jo-1 antigen (histidyl t-RNA synthetase).4–6 Anti-Jo-1 antibodies are associated with a characteristic clinical syndrome that may include muscle weakness and pain, Raynaud's phenomenon, interstitial lung disease, arthritis, and skin disorders. Pathological features in muscle also distinguish the Jo-1 syndrome from other types of immune myopathy.7 Characteristic histological features include prominent, but patchy, fragmentation and macrophage inflammation in perimysial connective tissue. Muscle fibre degeneration and regeneration are most marked in perifascicular regions neighbouring the connective tissue pathology.
Another myopathy syndrome includes rapidly developing and often severe weakness, and a serum “myositis specific” antibody directed against the signal recognition particle (SRP).5,8 The SRP, and its membrane associated receptor (SRPR or docking protein), catalyse the targeting of nascent secretory and membrane proteins to the protein translocation apparatus of the endoplasmic reticulum.9,10 The role of anti-SRP antibodies in the pathogenesis of the myopathy has not been defined. The anti-SRP associated syndrome has been described clinically in fewer than 20 patients.5,8 Pathological features of the myopathy have not been reported in detail. During the past three years we identified seven patients with myopathy syndromes and serum anti-SRP antibodies. We report the features of these patients, who have a characteristic clinical and pathological syndrome.
METHODS
Clinical data
Over a three year period, adult patients presenting to the neuromuscular service at Washington University School of Medicine in St Louis, Missouri, USA with acquired myopathies without other clear causes had serum evaluated for a panel of 12 myositis specific and myositis associated antibodies.
Assays were performed by the Oklahoma Medical Research Foundation (Oklahoma City, Oklahama, USA) using immunoprecipitation methodology. Assays included studies for antibodies to Jo-1, PL-7, PL-12, EJ, OJ, Mi-2, SRP, PM-Scl, Ku, U1RNP, U2RNP, and Ro. Results are reported qualitatively as positive or negative.
Our report includes the seven consecutive patients evaluated during this period in whom serum anti-SRP antibodies were detected and confirmed by serum immunoprecipitation and immunodiffusion methods. None of these patients had positive tests for antibodies to any of the other antigens studied. Anti-SRP antibodies were not detected in the serum of eight patients with dermatomyositis (five adults and three children) or in 31 patients with inflammatory myopathies tested during the same period.
Physicians in our neuromuscular group examined the seven patients with myopathy and anti-SRP antibodies. All had muscle biopsies that were evaluated in our neuromuscular pathology laboratory without knowledge of the antibody status of the patient. Clinical data were obtained from clinical examination and a review of the patients' case notes. Strength was evaluated on the MRC scale, and was also measured quantitatively, in six proximal and three distal muscle groups bilaterally, using a hand held myometer (Chatillon, Champion Scale, St Louis, Missouri, USA) and a grip meter (Jaymar, Preston, New Jersey, USA).11
We compared the myopathology in our seven patients with anti-SRP antibodies with muscle changes in 42 patients with no antibodies to SRP in their serum. Thirty six patients had definite polymyositis,12 definite dermatomyositis,12 or other subgroups of immune myopathies diagnosed on clinical, serological, electrophysiological, and pathological grounds.7,8 Specifically, for all these pathology control groups, we included only biopsies with myopathic, connective tissue, or vascular involvement. Serum anti-Jo-1 antibodies were confirmed using immunoprecipitation, as enzyme linked immunosorbent assay (ELISA) methodology alone can give false positive results. Six control patients had biopsies with no morphological abnormalities.
Laboratory data
Muscles chosen for biopsy had 4/5 strength (MRC grade). Five of the seven muscle biopsies were obtained before treatment with corticosteroids or immunosuppressive agents. The initial muscle biopsies from patients 2 and 5 were obtained two to four days after the onset of treatment with corticosteroids. Cryostat sections of rapidly frozen muscle were processed for muscle histochemistry and immunocytochemistry in a standard fashion.7,13 The evaluators (TM and AP) were blinded to the status of the anti-SRP antibodies at the time of initial biopsy evaluation and subsequent quantitative analysis. Immunocytochemistry was performed on biopsies from the seven patients with anti-SRP antibodies and paired normal tissue controls for each antibody using standard protocols. Primary antibodies used in this study were directed against CD68, CD3, and C5b-9 (membrane attack complex (MAC)) antigens (Sigma, St Louis, Missouri, USA) and human major histocompatibility complex class I antigen (MHC-1) (US Biological, Swampscott, Massachusetts, USA). Ulex europaeus agglutinin I (UEA-I) (Sigma), a lectin that binds to α-fucosyl residues on endothelial cells, was used as a marker for capillaries.14,15 Antibody binding was visualised using peroxidase conjugated secondary antibodies (Sigma).
Data analysis
Light microscopic images from regions randomly chosen for quantitative analysis were stored digitally. Endomysial vessel (capillary) density was determined by quantitating the number of endomysial vessels per 1000 μm2 of muscle fibre area (capillary index).14,15 The average endomysial capillary diameter was calculated from measurements of the smallest cross sectional diameters of endomysial vessels in randomly selected regions of digitally stored images of patient and control muscle biopsies. For analysis of C5b-9 deposition we evaluated only endomysial vessels, as antibodies to C5b-9 may stain the media of perimysial arterioles in normal muscle. Fisher's exact and Mann–Whitney rank sum tests were used to calculate the statistical significance of differences between groups.
CASE REPORTS
Patient 1
A 32 year old man was admitted to the hospital with a five month history of rapidly progressive severe weakness in the trunk, arms, and legs leading to inability to stand or sit. He had also noted generalised myalgia, fatigue, dyspnoea, and weight loss. Neurological examination showed bifacial weakness and severe symmetrical proximal weakness, graded at 0/5 in the lower extremities and 2/5 in the proximal upper extremities. Sensory and skin examination was normal. Laboratory data included raised creatine kinase (CK) at 6204 IU/l and raised aldolase at 55 IU/l. Electromyography revealed fibrillations, positive sharp waves, and myopathic motor unit potentials in proximal and intermediate muscles. Nerve conduction studies were normal except for a mildly reduced tibial CMAP amplitude. A muscle biopsy showed an active myopathy with muscle fibre degeneration and regeneration, markedly increased connective tissue, and a few small foci of mononuclear inflammatory cells. High dose intravenous corticosteroid treatment produced no improvement in strength.
Patient 2
A 35 year old woman developed weakness in the legs, and then in the arms, which progressed over 12 months. She also experienced myalgia, stiffness, and fatigue. Mild dysphagia was present initially but this did not persist. She was diagnosed as having polymyositis on clinical grounds, and treated using oral prednisone with symptomatic improvement in muscle strength. After two years, steroids were discontinued and the weakness and fatigue recurred. Reinstitution of prednisone and the addition of methotrexate and azathioprine were not effective. Over the next year, weakness progressed. Neurological examination showed symmetrical proximal weakness (3/5). Sensory and skin examination was normal. Laboratory data included a raised serum CK at 3064 IU/l. Electromyography revealed fibrillation potentials, positive sharp waves, and short duration, early recruited motor unit potentials in proximal muscles. Nerve conduction studies were normal. A muscle biopsy showed severe end stage pathology. Treatment with intravenous methylprednisolone (Solumedrol) (1 g/day for five days) followed by oral prednisone (60 mg/day for several months) produced mild improvement in functional abilities and proximal muscle strength.
Patient 3
A 48 year old woman developed weakness in the arms and legs and generalised fatigue that rapidly progressed over three months to severe disability with inability to rise from a bed or chair. She denied significant myalgia, skin changes, or bulbar or respiratory dysfunction. Neurological examination showed severe, relatively symmetrical proximal extremity muscle weakness (0/5 to 1/5) and mild neck flexor weakness (4/5), but no facial weakness. Serum CK was 24 900 IU/l. Electromyography revealed fibrillation potentials, positive sharp waves, and myopathic motor unit potentials in proximal muscles. A muscle biopsy showed an active myopathy with degenerating and regenerating muscle fibres, increased endomysial connective tissue, and no inflammation. The patient was treated with oral prednisone (60 mg/day) and azathioprine (50 mg/day) for two months with no change in symptoms. Drug doses were increased to 150 mg/day of azathioprine and 80 mg/day of prednisone. Quantitative muscle strength testing showed progressive improvement in proximal strength, reaching nearly normal values by 12 months, and continuing at those values over the next year with tapering doses of prednisone.
Patient 4
A 46 year old man developed weakness in the arms and legs that progressed over eight months to severe disability, with need for a walking frame and inability to rise from the sitting position. He also experienced diffuse myalgia, fatigue, and a 13.5 kg (30 pound) weight loss. General examination was notable only for mild tachycardia. Neurological examination showed severe symmetrical weakness, proximal greater than distal (2/5 to 3/5 in the legs and 0/5 in the proximal arms). Bilateral moderate facial and neck weakness was also present. Sensory and skin examination was normal. Serum CK was 8940 IU/l. Electromyography showed fibrillation potentials, positive sharp waves, and myopathic motor unit potentials in proximal muscles. Nerve conduction studies were generally normal. A muscle biopsy revealed an active myopathy, with degenerating and regenerating muscle fibres, but no inflammation. Treatment with oral prednisone (60 mg/day) for three months produced no improvement. Intravenous methylprednisolone (Solumedrol), 1 g/day for five days, followed by weekly single doses of 1 g, was associated with reduced fatigue and gradual improvement in strength and walking over 10 months.
Patient 5
A 39 year old woman presented with a two month history of progressive difficulty in climbing stairs and walking, falling, and a sense of generalised body heaviness. Serum CK was 7406 IU/l. Electrodiagnostic testing showed myopathic motor unit potentials in the biceps and early recruitment in the quadriceps muscles. Haematoxylin and eosin (H&E) stains of paraffin embedded muscle revealed scattered muscle fibre degeneration and regeneration but no inflammation. She was treated with a tapering dose of oral prednisone, beginning at 60 mg/day, and azathioprine 50 mg twice daily. Weakness continued to progress. Ten months after the onset of disease symptoms, general examination showed mild Cushingoid features and normal skin. On neurological examination there was symmetrical severe proximal weakness (0/5 to 1/5 in proximal arms and legs and 4/5 in the neck flexors). Facial strength was normal. Sensory examination was normal. Serum CK was 1950 IU/l. A second muscle biopsy showed an active myopathy with many degenerating and regenerating fibres and increased endomysial connective tissue. Treatment with weekly corticosteroids (0.75 to 1 g) for two years was associated with gradual and substantial improvement in strength, including 4/5 values in the proximal arms and legs and 5/5 in more distal muscles.
Patient 6
A 70 year old woman presented with a two month history of progressive proximal weakness, more in the legs than the arms, and generalised fatigue. A scaly skin rash was noted on her hands, neck, and eyelids. Serum CK was 25 000 IU/l. Treatment with oral prednisone, up to 80 mg/day, was followed by subjectively improved proximal strength. Tapering of the corticosteroid dose was associated with a moderate decline in proximal strength and increased disability. Neurological examination showed severe proximal weakness (0/5 to 3/5) in the arms and legs. Serum CK was mildly elevated at 473 IU/l. Electrodiagnostic testing was consistent with an irritable myopathy. A muscle biopsy revealed myopathic changes with many regenerating and some degenerating muscle fibres, but no inflammation. Increased doses of oral corticosteroids led to a subjective sense of increased strength.
Patient 7
A 66 year old woman presented with a one month history of progressive proximal weakness, exercise induced myalgia, a 2.3 kg (5 pound) weight loss, and mild dyspnoea on exertion. On examination there was severe proximal weakness (1/5 to 2/5). Serum CK was 15 034 IU/l. Electromyography showed fibrillation potentials and positive sharp waves with myopathic motor units in proximal and distal muscles. A muscle biopsy revealed an active myopathy with many degenerating and regenerating fibres but no inflammation. She was treated with corticosteroids, initially oral prednisone (60 mg/day), but when weakness progressed (to 0/5 in some muscles) this was changed to pulse dose intravenous, and then oral, methylprednisolone (Solumedrol). Methotrexate (15 mg/week) was also added. Improvement in strength and reduction in dyspnoea began after two months. Quantitative strength testing has shown mild progressive improvement in strength with residual severe weakness in proximal muscles.
RESULTS
Clinical characteristics of patients with anti-SRP antibodies
These data are shown in table 1. Our seven patients with myopathy and anti-SRP antibodies ranged in age from 32 to 70 years, with a mean of 48 years, at the onset of weakness. The onset of weakness in all patients was between August and January, usually in the autumn. Five patients were women.
Clinical and laboratory characteristics of the patients
All the patients had a history of proximal weakness that progressed relatively rapidly to severe disability. The time from onset to peak weakness ranged from two to 12 months, with a mean of five months. Six patients had a history of muscle discomfort, such as pain or fatigue, but this was never a primary complaint. Three patients noted dysphagia and two complained of dyspnoea. None had a history of palpitations or malignancy.
On examination all patients had proximal, mostly symmetrical, weakness involving both upper and lower limbs. Weakness was very severe in some proximal muscles in all patients. MRC grades in the weakest muscles ranged from 0/5 in five patients to a maximum of 3/5 (quantitative strength testing reduced to a maximum of 20% of normal) in two patients. The weakest muscles were always the deltoid and psoas. Very distal muscles, such as the intrinsic muscles of the hands and feet, were often only mildly weak or normal. Sensation was normal. One patient had a scaly rash that was not typical of dermatomyositis. After treatment with corticosteroids, six of the seven patients showed improvement on quantitative strength testing. Three patients relapsed, with increased weakness, during steroid tapering. Reintroduction of corticosteroid treatment did not result in the same level of improved strength as was observed initially.
Laboratory data
Laboratory data are shown in table 1. Serum CK was markedly raised at presentation in all seven patients with anti-SRP antibodies, ranging from 3064 to 25 000 IU/l (normal ≤ 170), with a mean of 12 900 IU/l. Aldolase was raised in the four patients tested, ranging from 23 to 174 IU/l (normal ≤ 8). Antinuclear antibodies were found in only one patient. The erythrocyte sedimentation rate (ESR) ranged from 1 to 42 (normal ≤ 30), and was mildly raised in four of the seven patients. Electromyography in all seven patients was consistent with a myopathy, showing small amplitude, brief, polyphasic motor unit potentials. Features of muscle irritability, including spontaneous fibrillation potentials and positive sharp waves, were very prominent.
Muscle pathology in patients with anti-SRP antibodies
These data are shown in tables 2 and 3 and fig 1.
Muscle pathology in patients with anti-SRP antibodies
Muscle pathology in immune myopathies: comparative features
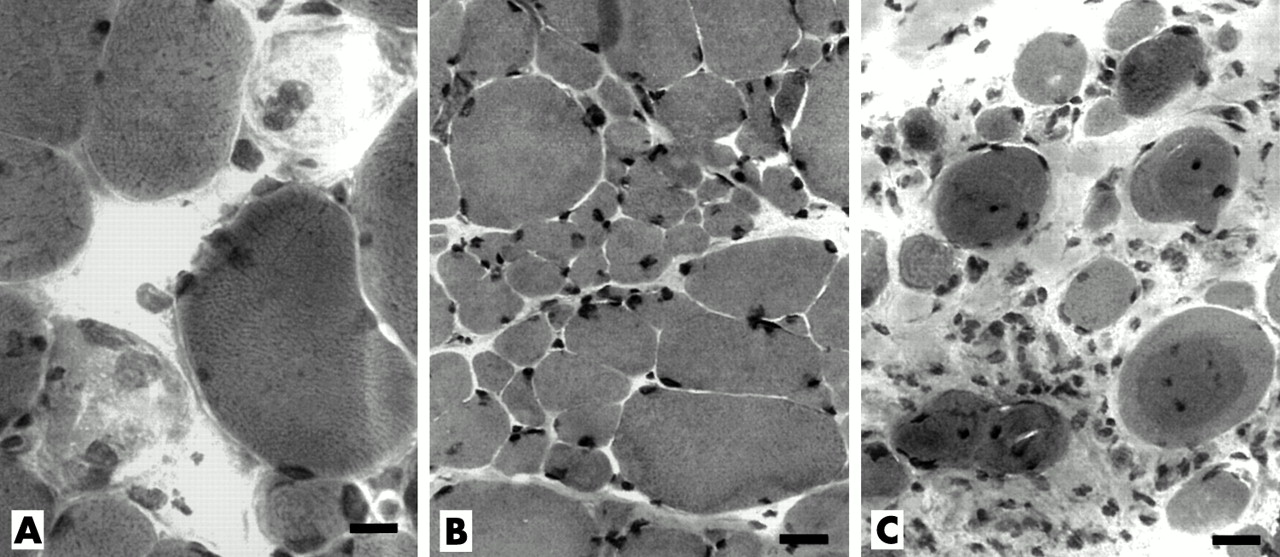
Muscle pathology in patients with myopathy and anti-SRP (signal recognition particle) antibodies. Early changes: (A) Muscle fibre necrosis. Scattered, pale, fragmented muscle fibres, some of which contain cells with large nuclei (macrophages). (Haematoxylin and eosin (H&E) stain, bar = 14 μM.) (B) Bimodal distribution of fibre sizes. The largest fibres are mildly hypertrophied. There is no inflammation. (H&E stain, bar = 18 μM.). Late changes: (C) Endomysial connective tissue is increased. Hypercontracted muscle fibres with abnormal internal architecture are present. (H&E stain, bar = 18 μM.)
One muscle biopsy (from patient 2) was end stage, with muscle fibres largely replaced by fat. The few remaining muscle fibres were small, rounded, and embedded in abundant connective tissue. Muscle biopsies from the other six patients with anti-SRP antibodies showed myopathic features including prominent variation of muscle fibre size (fig 1). None of the biopsies showed a perifascicular distribution of atrophic or myopathic changes.
The distribution of muscle fibre sizes often appeared bimodal. One population of muscle fibres was hypertrophied (fig 1, panels A and B). Many dark, rounded, probably hypercontracted, muscle fibres were also observed in two biopsies. The other population of muscle fibres was small, rounded, and often occurred in clusters (fig 1B). Small regenerating muscle fibres (basophilic colour on H&E stain, or intermediate colour on ATPase, pH 4.3) were distributed randomly through most biopsies.
Necrotic muscle fibres (diffuse pale staining and a hyaline appearance with H&E)—often invaded by macrophage-like cells that stained with non-specific esterase, acid phosphatase, and anti-CD68 antibody—were scattered through all six biopsies.
Prominently increased endomysial connective tissue was a feature of five biopsies (fig 1C). In several, the marked increase in connective tissue was especially notable in the light of the short (two to five month) history of weakness. In all biopsies NADH stains showed coarse, irregular internal architecture in many muscle fibres scattered throughout the biopsy.
Mononuclear inflammatory cells were uncommon. No biopsy had perimysial or perivascular accumulations of mononuclear cells. One biopsy showed a single focus of mononuclear cells in the endomysium. Focal cellular invasion of intact muscle fibres was not observed.
Alkaline phosphatase staining of perimysial connective tissue was noted in three patients. Mitochondrial stains reflected the abnormal internal architecture of muscle fibres that was also noted on NADH, but these were otherwise unremarkable. No biopsies had any muscle fibres with vacuoles. Staining of muscle fibres for MHC-I was absent in three biopsies, present only on scattered regenerating fibres in two, and present in scattered fascicles in small amounts in one.
Endomysial capillary changes in patients with anti-SRP antibodies
Endomysial capillary density was reduced in muscles from patients with anti-SRP antibodies (fig 2). The mean endomysial capillary index was reduced in muscles from patients with anti-SRP antibodies (p = 0.006), measuring 0.7 (0.1) (mean (SD)), compared with values in the normal controls of 1.2 (0.1). Muscle fibre profiles with no neighbouring capillaries were common (fig 2, panel E). Capillary density was uniformly reduced throughout the biopsies.

Endomysial capillary pathology. Normal muscle (panels A and C): numerous small endomysial capillaries are present; each muscle fibre cross section borders on several capillaries. (A, bar = 40 μM; C, bar = 8 μM.) Anti-SRP muscle (panels B, D, and E): endomysial capillaries are enlarged and reduced in number; muscle fibre cross sections may have no neighbouring capillaries. (B, bar = 40 μM; D and E, bar = 8 μM; UEA-I stain.)
In normal controls most muscle fibres had several neighbouring capillaries (fig 2, panels A and C). We have previously found—in agreement with other studies—that capillary density is normal in polymyositis and Jo-1 myositis, but reduced in dermatomyositis.7 The mean diameter of endomysial capillaries was increased in muscle from patients with anti-SRP antibodies (p = 0.001) (fig 2, panels B, D, and E), measuring 9.1 (0.3) μM compared with control values of 6.5 (0.4) μM (fig 2, panels A and C).
Membrane attack complex, the terminal C5b-9 components of complement, was deposited irregularly in endomysial capillaries in all seven biopsies (fig 3). C5b-9 staining was present in capillaries in most endomysial regions of anti-SRP muscles, in contrast to dermatomyositis in which deposition was usually patchy. No biopsy had diffuse staining of C5b-9 within muscle fibres. In one biopsy (patient 1) C5b-9 was deposited around the rim of scattered muscle fibres. Deposition of the terminal C5b-9 components of complement was not found in endomysial vessels in three control muscle biopsies without myopathy, or in the 19 patients with polymyositis.

{kind=link}
{kind=link}
{kind=link}
Terminal complement complex (C5b-9) in endomysial capillaries. C5b-9 is deposited in a patchy fashion in endothelial regions of scattered, enlarged endomysial capillaries (panel A). A high power image (panel B) shows C5b-9 deposition in four enlarged capillaries surrounding a single muscle fibre. A second high power image (panel C) illustrates the patchy nature of the C5b-9 deposition within the enlarged capillaries. (A, bar = 14 μM; B and C, bar = 7 μM). (D) Normal muscle shows no endomysial capillary staining for C5b-9 (bar = 12 μM).
DISCUSSION
Our study shows that myopathies associated with serum anti-SRP antibodies have characteristic clinical and pathological features. In previous studies anti-SRP antibodies have been associated with a myopathy syndrome which includes onset in the autumn and a rapidly developing severe weakness.5,8 Our results emphasise that severe weakness is a prominent clinical feature of the myopathy, occurring in all of our patients. The severity of weakness is often manifest as a complete loss of strength (0/5 on the MRC scale), which develops bilaterally in at least one muscle group. This occurred in five of our seven patients. In contrast, none of our 11 previously reported patients with myopathies associated with anti-Jo-1 antibodies7 had such severe weakness in any muscles (p = 0.003). Weakness in the anti-SRP antibody patients developed rapidly, with the time from onset to the maximum severity of weakness averaging only five months. The distribution of the weakness, which was symmetrical and predominantly proximal, was similar in all the patients with anti-SRP antibodies. After treatment with corticosteroids, weakness often improved, but only partially. Residual relatively disabling weakness in some muscle groups was the rule. As in other studies of individuals with anti-SRP antibodies,5,8 most of our patients complained of muscle discomfort, but the pain was never severe enough to require analgesic treatment. In contrast to previous series, none of our patients complained of palpitations or of other features suggesting cardiac involvement.
The onset of weakness in all our patients fell during the period between August and January. This differs from the onset of the anti-Jo-1 antibody related myopathy syndromes, which generally occurs in the spring.7 A seasonal pattern of disease onset might suggest an antecedent infectious or other environmental trigger to the anti-SRP antibody syndrome. However, no clear prodromal illness was identified in any of our patients.
A notable laboratory feature in all our patients, which probably reflected the active myopathic process, was the very high serum CK at presentation, ranging from 3064 to 25 000 IU/l, with a mean of 12 944. Serum aldolase was also raised in the four patients who were tested, ranging from 23 to 174 IU/l (normal less than 8). The EMG suggested an active myopathy, showing myopathic motor unit potentials and prominent spontaneous activity in proximal muscles in all the patients. There were few laboratory signs of systemic disease in our patients: the ESR ranged from 1 to 42, with a mean of 27, while antinuclear antibodies were mildly raised in only one patient.
Several different pathological patterns occur in immune and inflammatory myopathies.16,17 In some polymyositis syndromes mononuclear cell infiltration of the endomysium and focal cellular invasion of muscle fibres are the rule. Specific pathological features may be more common in subgroups of inflammatory or immune myopathies. In Jo-1 antibody associated myositis a common feature is inflammation in, and fragmentation of, perimysial connective tissue.7 Notable features in other syndromes include cytochrome oxidase negative muscle fibres in a group of patients with polymyositis unresponsive to corticosteroid treatment,18 and inclusion bodies in inclusion body myositis.19 In dermatomyositis and Jo-1 antibody associated myositis, a perifascicular localisation of muscle fibre pathology is common. Perivascular inflammation may be more prominent when immune myopathies occur in the setting of a systemic connective tissue disease,20 but can also be found in disorders with no clear immune pathogenesis, such as inclusion body myositis and some muscular dystrophies.
Capillary pathology somewhat similar to that in our patients with myopathy and anti-SRP antibodies has been described in dermatomyositis15 and in necrotising myopathy with pipestem capillaries.21 Deposition of the complement component C5b-9 (membrane attack complex) in capillaries, a reduction in the capillary density (capillary index), and enlargement of endomysial capillaries is common to the anti-SRP syndrome and those other two disorders. Enlarged capillaries with thickened PAS positive walls and small lumens—as described in the pipestem capillary syndrome—were not present in muscles from patients with anti-SRP antibodies.
The capillary pathology in the anti-SRP antibody syndrome with C5b-9 deposition and enlarged lumens and outside diameters is similar to the changes reported in dermatomyositis. However, the capillary pathology, enlarged size, and C5b-9 deposition in the anti-SRP antibody syndrome occurred diffusely throughout the muscle rather than having a patchy distribution as is typical of dermatomyositis.22 Other differences from dermatomyositis in patients with anti-SRP antibody associated myopathies included the severe diffuse weakness, absence of rash, and, in the muscle biopsy, prominently increased endomysial connective tissue and relatively enlarged muscle fibres that often appeared hypertrophied or hypercontracted. Further, none of our control patients with typical dermatomyositis syndromes had serum anti-SRP antibodies.
Clinical, laboratory, and pathological features of the anti-SRP syndrome also distinguish it from other immune myopathy syndromes (table 4). There are few of the systemic features such as skin disorders, arthritis, interstitial lung disease, or associated neoplasms that occur in other myositis syndromes. The anti-SRP syndrome—with rapidly progressive severe proximal weakness, occasionally associated with prominent weight loss—has clinical similarities to paraneoplastic necrotic myopathies.13 However, the syndromes differ in their pathology. In paraneoplastic necrotic myopathies, large regions of necrotic C5b-9 positive muscle fibres are typical features,23 while prominently increased endomysial connective tissue or enlarged capillaries are not usually present (table 3). This differs from the muscle pathology in the anti-SRP syndrome, with its appearance of a chronic active myopathy, including prominent endomysial connective tissue and scattered necrotic muscle fibres that are only rarely positive for C5b-9. The explanation for the prominently increased connective tissue in the anti-SRP syndrome might be inhibition of muscle fibre regeneration, or stimulation of fibroblasts by antibodies, as is found in systemic sclerosis24 or fibrogenic cytokines.25
Comparative features in selected immune myopathy syndromes
Another common feature of anti-SRP myopathies that differs from many immune or inflammatory myopathies is the rarity, or absence, of foci of mononuclear inflammatory cells. Other than the CD68 and acid phosphatase positive cells associated with necrotic muscle fibres, anti-SRP biopsies show few or no foci of perimysial or endomysial mononuclear cells, and no focal invasion of muscle fibres by mononuclear cells. Groups of CD3 positive inflammatory cells were only detected in one patient (patient 1). In contrast to inclusion body myositis and some types of polymyositis,26 MHC-I antigen was not prominently expressed on muscle fibres, except in scattered regenerating fibres, in most of the biopsies.
The improvement after corticosteroid treatment in patients with myopathies and anti-SRP antibodies suggests that the syndrome may be immune mediated. Several features in muscle biopsies from patients with anti-SRP antibodies support the idea that immune mechanisms, especially humoral factors, may play a role in the pathogenesis of the myopathy.
First, perimysial connective tissue showed staining for alkaline phosphatase in biopsies from three anti-SRP patients. In our experience, staining of perimysial connective tissue for alkaline phosphatase occurs commonly in immune mediated myopathies and rarely in muscular dystrophies or inclusion body myositis.7
Second, the most consistent pathological feature suggesting the presence of a humoral immune disorder is the pattern of non-inflammatory capillary pathology in all anti-SRP patients. This pathological pattern is similar to that found in dermatomyositis, a disorder in which humoral immune mechanisms are thought to play a prominent role. The pattern of prominent vascular pathology could suggest that muscle fibre damage in anti-SRP antibody associated myopathies is related to ischaemia. The frequent finding of myopathic grouping—multiple foci of small rounded, muscle fibres that often appear to be immature or regenerating—might support the idea of a multifocal ischaemic pathology.
The relation of antibody binding to SRP, an intracellular antigen, to the pathogenesis of the associated myopathy syndrome is uncertain. It has been difficult to show that antibodies directed against intracellular antigens play a role in any neuromuscular disorder.27 SRP is a ubiquitous protein complex in eukaryotic organisms that docks newly translated proteins at the endoplasmic reticulum.9,10 This function involves binding of a hydrophobic signal sequence on nascent proteins and targeting of the ribosome nascent protein–SRP complex to the membrane through a GTP dependent interaction with the SRP receptor. It will be interesting to study whether anti-SRP antibodies, or other serum factors, from our patients with myopathies are capable of interfering with any phase of this process in cells, either in general or in muscle fibres in particular.
Overall, characteristic clinical features of myopathies associated with anti-SRP antibodies include rapid progression of weakness over a period of months, culminating in severe disability. The anti-SRP syndrome may be more common than previously recognised. We identified seven patients with anti-SRP antibodies in a three year period, from a total of 95 patients diagnosed in our clinical and pathological service with immune or inflammatory myopathies. Further, the anti-SRP antibody has been detected in 14% of European patients with immune mediated myopathies.4 Pathologically, the myopathy shows active muscle fibre degeneration and regeneration and a prominent increase in endomysial connective tissue, but little or no mononuclear cell inflammation. The pathological features suggesting an immune pathogenesis, including small vessels, with altered morphology and complement deposition, and perimysial connective tissue, with alkaline phosphatase staining, may be missed on routine muscle histochemistry. Clinically, the myopathy with anti-SRP antibodies should be included in the differential diagnosis of syndromes with rapidly progressive weakness and a high serum CK, especially when the muscle biopsy shows some of the characteristic pathological features described here. Strength may improve when corticosteroid treatment is given early. However, confirmation of appropriate treatment strategies will require further study.
REFERENCES
Footnotes
-
Competing interests: none declared