Article Text

Abstract
Objective Systemic sclerosis-associated pulmonary arterial hypertension differs from idiopathic pulmonary arterial hypertension with respect to histopathology, treatment responses and survival. Medical progress on PAH is hampered by the lack of human biosamples and suitable animal models. In this study, the authors evaluated fos-related antigen 2 (Fra-2) transgenic mice as a novel model for systemic sclerosis-associated pulmonary arterial hypertension.
Methods Lung sections of Fra-2 transgenic (n=12) and wild-type mice (n=6) were analysed at 16 weeks by histology using Dana Point criteria. Cellular and molecular key players were assessed by immunohistochemistry. To test the model's sensitivity to change over treatment, a subgroup of Fra-2 transgenic mice (n=6) was treated with the tyrosine kinase inhibitor nilotinib twice daily 37.5 mg orally from 8 weeks of age.
Results Fra-2 transgenic mice developed severe vascular remodelling of pulmonary arteries and non-specific interstitial pneumonia-like interstitial lung disease resembling human systemic sclerosis-associated pulmonary hypertension. Histological features typical for systemic sclerosis-associated pulmonary arterial hypertension, such as intimal thickening with concentric laminar lesions, medial hypertrophy, perivascular inflammatory infiltrates, adventitial fibrosis, but not pulmonary occlusive venopathy were frequently detected. Platelet-derived growth factor signalling pathways were activated in pulmonary vessels of Fra-2 transgenic compared with wild-type mice. Since treatment with nilotinib strongly prevented the development of proliferative vasculopathy and lung fibrosis, the model proved to be sensitive to treatment.
Conclusions This study suggests that Fra-2 transgenic mice as an animal model of systemic sclerosis-associated pulmonary arterial hypertension display main characteristic features of the human disease. It therefore allows studying pathophysiological aspects and might serve as a preclinical model for interventional proof-of-concept studies.
Statistics from Altmetric.com
Pulmonary arterial hypertension (PAH) and interstitial lung disease (ILD) account for more than 60% of the systemic sclerosis (SSc)-related mortality till date.1
Patients with SSc-PAH have a worse prognosis and response to PAH-specific therapies than patients with idiopathic PAH (IPAH) or PAH related to other connective tissue diseases.2,–,4 The observed differences in SSc-PAH and the frequent co-existence of ILD5 support the concept of a specific pathophysiology of SSc-related pulmonary hypertension (PH), and two recent studies suggested remarkable histopathological differences of the vascular lesions associated with IPAH and those related to SSc-PAH.6 ,7
Since research on PAH is hampered by the lack of human biosamples, animal models are of utmost importance (1) to study pathophysiological interactions between the different lung manifestations, (2) to identify molecular key players and potential therapeutic targets, and (3) for preclinical proof-of-concept studies. Unfortunately, there is a shortage of validated animal models of SSc that simultaneously display features of ILD and pulmonary vasculopathy,8 whereas established animal models of PAH rather reflect features of human IPAH than SSc-PAH.9
The Fra-2 (fos-related antigen-2) transgenic (tg) mouse model combines vasculopathy with fibrosis of the skin and internal organs, and Fra-2 protein is overexpressed in the skin and lungs of patients with SSc.10 ,11 The aim of our study was to analyse Fra-2 tg mice as a potential animal model of SSc-PH1 by exploring whether it displays the histopathological features considered specific for SSc-PAH,2 by characterising key cellular and molecular pathways contributing to the SSc-PAH-specific features and3 by assessing its sensitivity to change over treatment.
Materials and methods
Additional information on methods is provided in the online supplementary material.
Animals
A subgroup of Fra-2 tg mice10 ,11 (n=6) was treated with nilotinib 2×37.5 mg/d and compared with vehicle-treated Fra-2 tg mice and wild-type (wt) littermates (n=6 each). Fra-2 tg mice were backcrossed from a mixed (C57BL/6×CBA) genetic background on a pure C57/Bl6 background for at least six generations.11
Histology
Lung sections were prepared as described previously.12 Sections were stained with H&E and Masson's trichrome staining according to standard protocols.
Immunohistochemistry
For primary and secondary antibodies, refer to the online supplementary material.
Analysis of histological and immunohistochemical staining
All slides were analysed by two blinded independent examiners. Pictures were taken with a digital camera on an Imager1 microscope (Carl-Zeiss AG, Feldbach, Switzerland), using AxioVision software Release V.4.6.
Pulmonary histopathology was assessed by microscopic criteria including pattern and distribution of inflammation, deposition of extracellular matrix and architectural changes.13 Vascular histopathology was analysed according to the Dana Point consensus criteria.14
Vascular remodelling was assessed by calculating the median(Q1,Q3) of vessel wall thickness and the percentage of luminal occlusion of pulmonary arteries using GraphPad Prism software.
Cellular key players were identified by double staining with proliferation cell nuclear antigen (PCNA) as proliferation marker and the respective cell markers for vascular smooth muscle cells (α-smooth muscle antigen (α-SMA)+ smooth muscle22α (SM22α)+) and myofibroblasts (α-SMA+). Manually counted positive nuclei within vessel walls were confirmed independently by automated counting using image analysis software (Image J, NIH).
To assess differences in the expression of platelet-derived growth factor (PDGF)-BB and p(phosphorylated)-PDGF receptor β (PDGFRβ), positively stained vascular cells were analysed by automated analysis of staining intensity (0=no, 1=weak, 2=moderate and 3=intensive staining) using Image J. The median(Q1,Q3) of staining intensity was calculated using GraphPad Prism software.
For the analysis of inflammatory infiltrates, stains for T cells (CD3+) and murine macrophages (F4/80+) and double stainings with PDGF-BB/p-PDGFRβ were performed.
Statistical analysis
Non-parametric non-related data were analysed with the Mann–Whitney U test and expressed as median(Q1,Q3). p Values <0.05 were considered statistically significant. Power calculation was performed using STATA V. 10.0 (StataCorp, College Station, Texas).
Results
Pulmonary pathology of Fra-2 tg mice resembles changes in SSc-PH
Pulmonary vasculopathy
To investigate Fra-2 tg mice as a potential model for SSc-PAH, we analysed features of pulmonary vascular remodelling.
Increase in wall thickness and occlusion of pulmonary arteries were the most prominent features of pulmonary pathology in Fra-2 tg (figure 1B) compared with wt mice (figure 1A). Semi-quantitative analysis of vascular remodelling showed that in Fra-2 tg mice the thickness of vessel walls was strongly increased compared with wt mice (median(Q1,Q3) 44(32,59)μm vs 21.5(13,36)μm; p<0.0001) (figure 1C). Obliterated vessels were almost undetectable in wt mice (0(0,1)%), whereas the percentage of obliterated vessels was increased to 33(33,53)% in Fra-2 tg mice (p=0.04) (figure 1D).

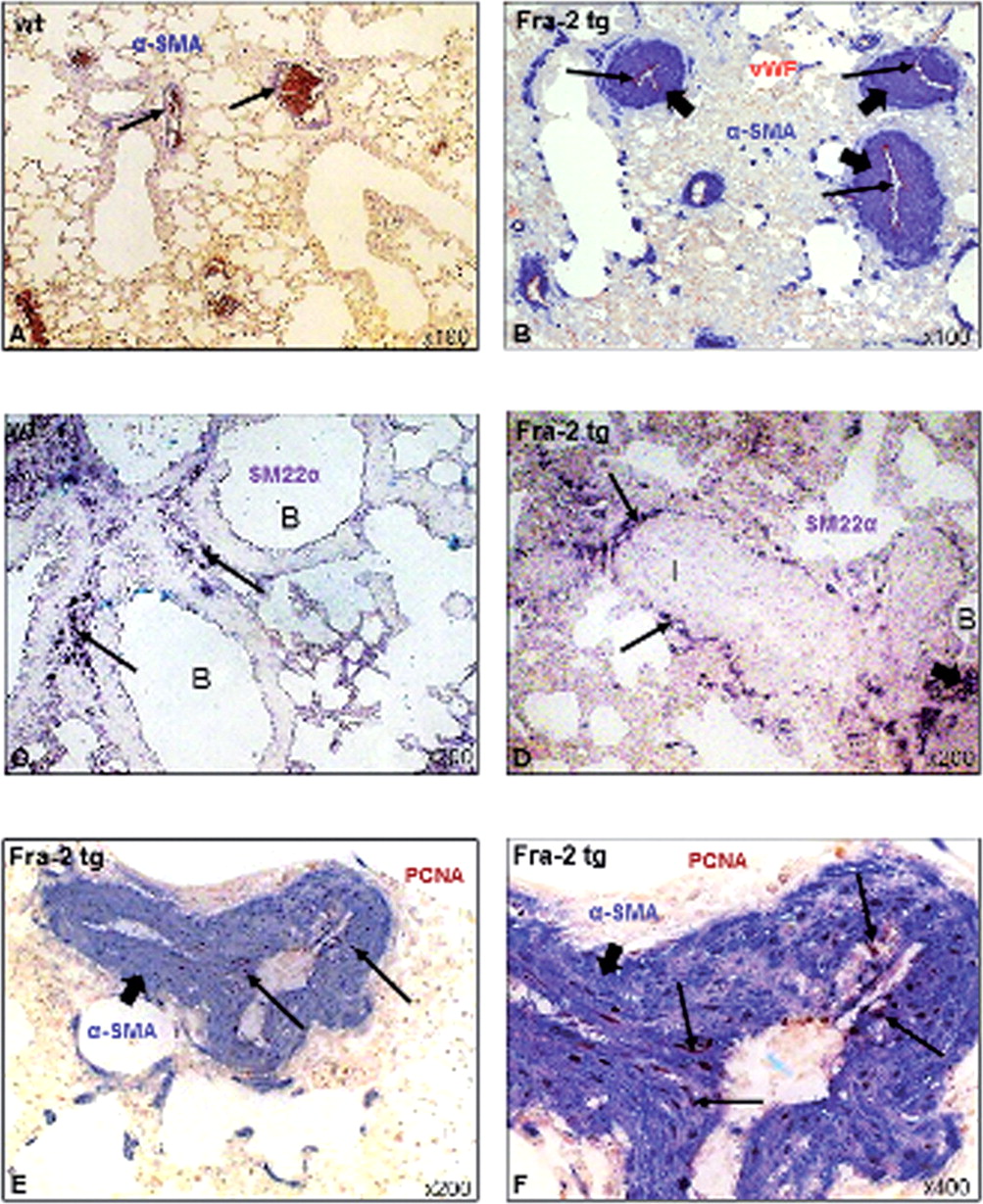
Pulmonary pathology of Fra-2 tg mice: vascular remodelling (A) Pulmonary histology of wt mice (H&E staining, arrow indicates vascular structure, B indicates bronchus). (B) Remodelling of pulmonary vessels with luminal narrowing and obliteration in Fra-2 tg mice (arrows; H&E staining, B indicates bronchi). (C) and (D) show the semi-quantitative analysis with evaluation of vessel wall thickness and luminal obliteration in Fra-2 tg compared with wt mice. (E–H) illustrate the main pathological changes of pulmonary vessels in Fra-2 tg mice: concentric laminar lesion due to formation of neointima by myofibroblasts and VSMCs (E, α-SMA positive, blue, short arrows) whereas the endothelial cell layer (vWF positive, red, long arrows) remained unaffected; (F) medial hypertrophy (arrow, H&E staining) and perivascular inflammatory infiltrates (white asterisk), (G) adventitial fibrosis (Masson's trichrome stain, bold arrow) and concentric laminar lesion (long arrow), and (H) eccentric non-laminar lesion (Masson's trichrome stain, arrow) due to expansion of fibroblasts and deposition of extracellular matrix proteins. Pictures are representative examples of 6 wt and 6 Fra-2 tg mice. Data are expressed as median and interquartile range. * indicates p values <0.05. Fra-2, fos-related antigen 2; α-SMA, α-smooth muscle antigen; tg, transgenic; VSMC, vascular smooth muscle cells; vWF, van Willebrand factor; wt, wild-type.
We next evaluated the histopathology of pulmonary vessels according to the Dana Point criteria14 (table 1).
Pathological characteristics of vasculopathies in pulmonary hypertension
In summary, Fra-2 tg mice developed several features that are considered more common in SSc-PAH than in IPAH. In contrast to human SSc-PH, pulmonary occlusive venopathy (POV, formerly PVOD) was not detectable in Fra-2 tg mice.
Interstitial lung disease
Given the frequent co-existence of ILD, we additionally examined the histology of interstitial lung changes (for details, see the online supplementary material).
In summary, Fra-2 tg mice developed a severe interstitial pneumonitis and fibrosis with concomitant lung emphysema (online supplementary figure 1) which closely resembled the features of human non-specific interstitial pneumonia (NSIP), which is the main histological pattern in SSc-ILD.15 In contrast to a previous study,10 honeycombing, severe scarring or other pulmonary changes resembling human UIP (usual interstitial pneumonia) were not observed which might be due to the different genetic background of the Fra-2 tg mice.
Cellular composition of intimal lesions
We next characterised the cells driving the intimal thickening in Fra-2 tg mice. In wt mice, α-SMA staining indicated muscular pulmonary vessels (figure 2A). α-SMA+ myofibroblasts/vascular smooth muscle cells (VSMCs) (figure 2B) were the most abundant cells within the neointima, whereas expansion of endothelial cells (van Willebrand factor (vWF)+) was not observed (figure 2B). To differentiate between VSMCs and myofibroblasts, we performed additional stainings with antibodies against the VSMC-specific marker SM22α (figure 2C, D). In wt mice, VSMCs were mainly found within the walls of bronchi (figure 2C), in Fra-2 tg mice, mainly in the media but not the intima of pulmonary vessels and in bronchiolar walls (figure 2D).

Cellular key players of vascular remodelling in Fra-2 tg mice. Compared with wt mice (A), severe vascular remodelling of pulmonary arteries could be observed in Fra-2 tg mice (B). In Fra-2 tg mice, increase in vessel wall thickness occurred due to expansion of α-SMA positive cells (dark blue, short arrows), whereas the endothelial cell layer was not increased (vWF-positive, red, long arrows). Increase in vessel wall thickness was mainly due to myofibroblasts (α-SMA positive, SM22α negative), whereas VSMCs (α-SMA (blue) and SM22α (purple) positive) were mainly expressed in bronchiolar (B) walls in wt mice (C, arrows), and additionally in the media of muscular vessels in Fra-2 tg mice (D, arrows). (E) Formation of neointima was mainly due to proliferation of α-SMA-positive cells (dark blue, short arrows) as indicated by double staining with the proliferation marker PCNA (brown, long arrows). (F) shows the proliferating cells in a higher magnification. Pictures are representative examples of 6 wt and 6 Fra-2 tg mice. Fra-2, fos-related antigen 2; PCNA, proliferation cell nuclear antigen; α-SMA, α-smooth muscle antigen; SM22α, smooth muscle 22; tg, transgenic; VSMC, vascular smooth muscle cells; wt, wild-type.
The proliferation marker proliferating cell nuclear antigen (PCNA) (figure 2E,F) was abundantly expressed by myofibroblasts showing that the observed changes were due to proliferation and not hypertrophy of cells.
In summary, mainly myofibroblasts, and to a lesser extent VSMCs, were key players in the remodelling of pulmonary arteries in Fra-2 tg mice which differs from the previous finding of VSMCs as the most abundant cells. This difference might either be due to technical reasons (no additional staining with a VSMC-specific marker, such as SM22α+), or due to the different genetic background of the Fra-2 tg mice.
Molecular mechanisms of vascular remodelling: PDGF signalling
Next, we assessed potential molecular mechanisms of both vasculopathy and ILD. In patients with IPAH, PDGF-BB has been proposed as a novel therapeutic target,16 ,17 and a recent study reported a higher immunoreactivity for p-PDGFRβ in SSc-PAH compared with IPAH.18
Thus, we analysed the expression of PDGF-BB and the phosphorylated (=activated) PDGFRβ in the lungs of Fra-2 tg and wt mice by assessing the staining intensity. Both, the expression of PDGF-BB (figure 3B) and of p-PDGFRβ (figure 3E) was increased in vascular structures of Fra-2 tg compared with wt mice (figure 3A,D). Occasionally, increased staining for p-PDGFRβ could be observed in the alveolar epithelium of Fra-2 tg mice (figure 3E). Semi-quantitative analysis confirmed the substantially increased expression of PDGF-BB in pulmonary vessels in Fra-2 tg compared with wt mice (median(Q1,Q3) of staining intensity 3(2.8,3.0) vs 0(0,1); p<0.0001) (figure 3C). A similarly strong upregulation was observed for p-PDGFRβ (3(2,3) vs 0(0.0,0.3); p<0.0001) (figure 3F). In addition to vascular structures, tissue macrophages (F4/80+) in Fra-2 tg mice showed a strong upregulation of both PDGF-BB (figure 3G) and p-PDGFRβ (figure 3H). These findings suggest a potential role for PDGF-BB in the pulmonary pathophysiology of Fra-2 tg mice.
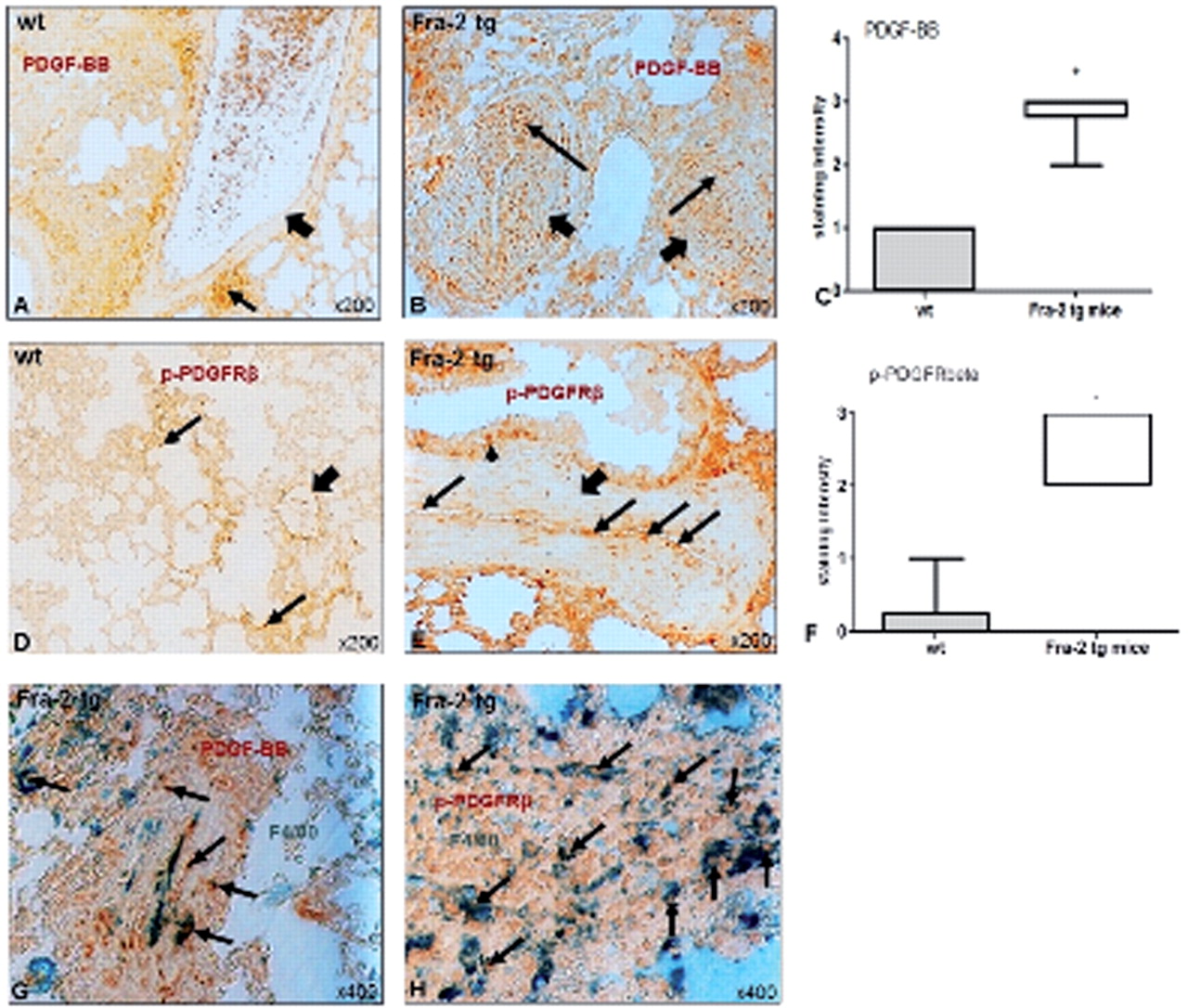
Molecular mechanisms of vascular remodelling in Fra-2 tg mice: PDGF signalling. Compared with wt mice (A), in Fra-2 tg mice (B), vascular structures (short arrows) showed an increased staining for PDGF-BB (brown, long arrows) as shown in the semi-quantitative analysis (C). The same difference could be observed for phosphorylated PDGFRβ (brown, long arrows; arrowhead indicates increased staining in the alveolar epithelium) in pulmonary vessels (bold arrow) of Fra-2 tg (E) compared with wt mice (D; bold arrow indicates vascular structure, arrows indicate staining in the interstitium). (F) shows the respective semi-quantitative analysis. In addition, there was also an increased expression of PDGF-BB (G, brown) and its activated receptor (H, brown) in pulmonary macrophages (green, F4/80-positive, arrows). Pictures are representative examples of 6 wt and 6 Fra-2 tg mice. Data are expressed as median and interquartile range. * indicates p values <0.05. Fra-2, fos-related antigen 2; PDGF, platelet-derived growth factor; PDGFR, PDGF receptor; tg, transgenic; wt, wild-type.
Nilotinib prevents vascular remodelling and fibrosis in Fra-2 tg mice
Next we evaluated whether targeting the PDGF-BB/PDGFR pathway using the tyrosine kinase inhibitor nilotinib could modify the vascular and fibrotic lesions in Fra-2 tg mice.
Pulmonary vasculopathy
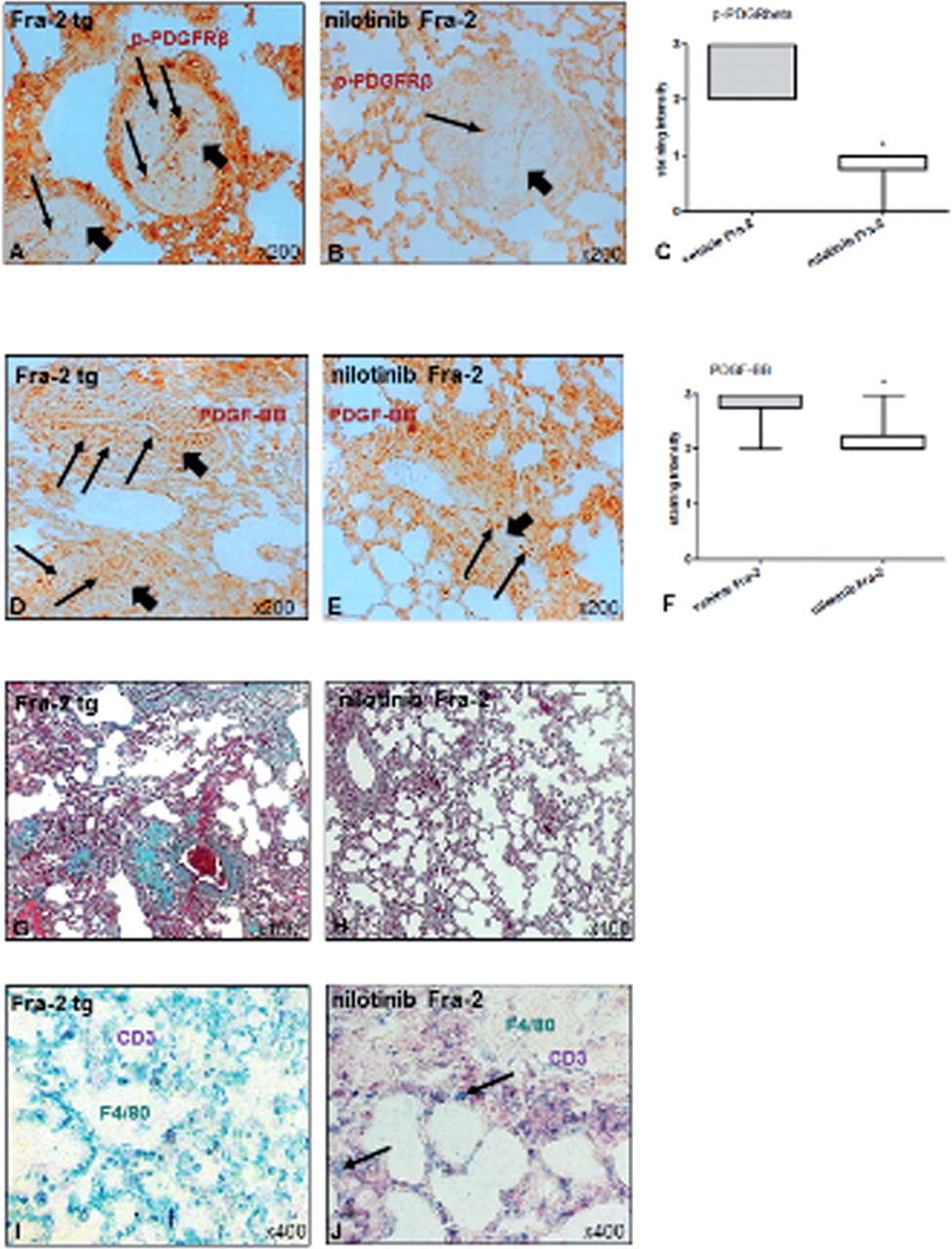
Treatment with nilotinib strongly prevented the remodelling of pulmonary arteries (figure 4B) compared with vehicle-treated Fra-2 tg mice (figure 4A). The endothelial cell layer (vWF+) was not affected upon treatment with nilotinib (figure 4B) compared with wt mice (figure 4A). Semi-quantitative analysis showed that compared with vehicle-treated Fra-2 tg mice, in nilotinib-treated Fra-2 tg mice both the thickness of vessel walls (median(Q1,Q3) 44(32,59) vs 22.5(18,28)μm, p <0.0001) (figure 4C) and the percentage of obliterated vessels (33(13,53)% vs 0(0,7)%, p=0.03) (figure 4D) was significantly reduced reaching the levels of wt mice.

Effects of nilotinib on vascular remodelling in Fra-2 tg mice. Compared with vehicle-treated mice (A, short arrows), treatment with nilotinib strongly prevented the vascular remodelling of pulmonary arteries (B, short arrows). The endothelial cell layer was not affected (red, vWF positive cells, long arrows. (C and D) demonstrate the differences in vessel wall thickness and luminal obliteration in the semi-quantitative analysis. As indicated by the double staining with the proliferation marker PCNA (brown, long arrows) compared to vehicle-treated mice (E), the expansion of α-SMA-positive cells (blue, myofibroblasts and VSMCs, short arrows) was reduced upon treatment with nilotinib (F), additionally demonstrated in the semi-quantitative analysis (G). Pictures are representative examples of 6 wt, 6 vehicle-treated and 6 nilotinib-treated Fra-2 tg mice. Data are expressed as median and interquartile range. * indicates p values <0.05. Fra-2, fos-related antigen 2; PCNA, proliferation cell nuclear antigen; α-SMA, α-smooth muscle antigen; tg, transgenic; VSMC, vascular smooth muscle cells; vWF, van Willebrand factor; wt, wild-type.
Treatment with nilotinib resulted in a reduced proliferation of α-SMA+cells compared with vehicle-treated Fra-2 tg mice as analysed by PCNA-double-staining (figure 4F,E). These findings were confirmed by semi-quantitative analysis with reduction of proliferating (PCNA+) α-SMA+cells in nilotinib-treated compared with vehicle-treated Fra-2 tg mice (median(Q1,Q3) 22(19,43) vs 87(57,90) PCNA+vascular cells/high-power field) (figure 4G).
As expected, the expression of p-PDGFRβ (figure 5B), and also of PDGF-BB (figure 5E) was reduced in vascular structures compared with vehicle-treated mice (figure 5A,D) showing successful targeting of PPDGFR. Semi-quantitative analysis demonstrated a reduced staining intensity for p-PDGFRβ (figure 5C) and also for PDGF-BB (figure 5F) in the pulmonary vessels of nilotinib-treated compared with vehicle-treated Fra-2 tg mice (median(Q1,Q3) 2(2.0,2.3) vs 3(2.8,3.0), p=0.03; 1(0.8,1.0) vs 3,(2,3) p=0.004).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effects of nilotinib on PDGF signalling and interstitial lung disease. Compared with vehicle-treated Fra-2 tg mice (A, D), the expression of both p-PDGFRβ (B, brown, long arrows) in vascular structures (short arrows) and PDGF-BB (E, brown, long arrows) was reduced by treatment with nilotinib. (C and F) show the results of the respective semi-quantitative analyses. Interstitial fibrosis (appears green in Masson's trichrome staining) (H) was strongly prevented by nilotinib compared with vehicle-treated Fra-2 tg mice (G). In 3 out of 6 mice, nilotinib (J) diminished the number of macrophages (F4/80 positive, green, arrows) compared with vehicle-treated mice (I) whereas CD3 positive cells (purple) remained unaffected. Pictures are representative examples of 6 wt, 6 vehicle-treated and 6 nilotinib-treated Fra-2 tg mice. Data are expressed as median and interquartile range. * indicates p values <0.05. Fra-2, fos-related antigen 2; PDGF, platelet-derived growth factor; PDGFR, PDGF receptor; tg, transgenic; wt, wild-type.
Interstitial lung disease
Treatment with nilotinib also inhibited the development of lung fibrosis in Fra-2 tg mice when assessing the accumulation of connective tissue by Masson's trichrome staining (figure 5G,H).
Interestingly, treatment with nilotinib diminished the numbers of macrophages (figure 5J, F4/80+), whereas T cells (figure 5J, CD3+) were not affected compared with vehicle-treated Fra-2 tg mice (figure 5K). However, there was a considerable heterogeneity between mice in the effects of nilotinib on inflammatory cells, and the differences to vehicle-treated mice, therefore, did not reach statistical significance in the semi-quantitative analysis.
Discussion
To date, there is substantial evidence on several levels that SSc-PAH, though sharing similarities, substantially differs from IPAH and thus might warrant different therapeutic approaches. However, the development of targeted therapies is hampered by the lack of human biosamples of SSc patients and by the lack of suitable animal models for preclinical studies.
Our study suggests Fra-2 tg mice as a potential model for SSc-PAH. The comprehensive histological and immunohistochemical analysis showed that Fra-2 tg mice displayed many pathological changes characteristic for the vascular remodelling in human SSc-PAH.6 ,7 ,18 However, the model cannot be used to further delineate mechanisms underlying POV, as POV-specific features were not identified in our study. Besides vasculopathy, interstitial inflammation and fibrosis closely resembling human NSIP as the most common form of SSc-ILD could be observed. Fibroblastic foci and honeycombing, associated with UIP, were rarely detectable in our study, but were identified more frequently in a recent study.10 This might be explained by the different background of Fra-2 tg mice, as the mice in this study were backcrossed from a mixed (C57BL/6×CBA) genetic background on a pure C57/Bl6 background.
However, besides allowing the evaluation of pathophysiological mechanisms and, in particular, the interplay of pro-fibrotic and vascular processes, Fra-2 tg mice might also serve as a preclinical model for interventional proof-of-concept studies. The tyrosine kinase inhibitor nilotinib by blocking PDGFR signalling prevented the development of both proliferative pulmonary vasculopathy and fibrosis in Fra-2 tg mice which argues for SSc-specific therapies of PH. Furthermore, our data support previous studies on PDGF as key mediator of vasculopathy in SSc-P(A)H.17,–,19 Inhibition of PDGF signalling, for example, by tyrosine kinase inhibitors, might be an example of a promising future therapy for P(A)H20 with particular evidence in SSc-PAH, as it targets vascular and fibrotic changes.21 ,22
The role of Fra-2 as a potential key player for the development of (pulmonary) vasculopathy and fibrosis in SSc should be addressed in further studies. Recent data demonstrated an increased expression of Fra-2 in lung biopsies from patients with UIP, NSIP and SSc-associated NSIP.10 Based on previous data on microvasculopathy and dermal fibrosis in SSc patients,11 ,23 a recent study demonstrated substantial antifibrotic effects of AP-1 (activator protein-1) inhibition in different animal models of SSc.24 Thus, Fra-2/AP-1 itself might represent an interesting molecular target for future SSc-specific therapies, even more so, since several pro-inflammatory chemokines and cytokines implicated in the pathogenesis of SSc were expressed in high levels in the lungs of Fra-2 tg mice including CXCL5 (chemokine (C-X-C motif) ligand 5), CCL2 (chemokine (C-L motif) ligand 2), IL-2 (interleukin-2), IL-4 and IL-6.10 Inhibition of these downstream targets with currently available drugs might elucidate further treatment options. AP-1 is induced by growth factors, cytokines and oncoproteins which are involved in the proliferation, survival, differentiation and transformation of cells.25 Whether Fra-2-related pathogenic changes in vivo might be accelerated by other factors, such as pro-inflammatory stimuli or hypoxia26 in pulmonary disease, will have to be addressed in future studies.
Taken together, the model of Fra-2 tg mice is the first animal model that simultaneously displays major histopathological features of pulmonary SSc, for example, pulmonary proliferative vasculopathy and interstitial fibrosis, which occur frequently together in patients with SSc and are associated with high mortality. Fra-2 tg mice hold great promise to further delineate the pathophysiological links between vascular remodelling and fibrosis in pulmonary SSc and to identify potential specific molecular and cellular targets for intervention. This study also underlines a prominent role of PDGF in the pathophysiology of pulmonary SSc and suggests Fra-2 tg mice as a preclinical model for interventional proof-of-concept studies in pulmonary SSc.
Acknowledgments
The authors thank Maria Comazzi for the excellent technical support.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Web Only Data - This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Funding BM received the Encysive Young Investigators Award; AJ received the IAR-EPALINGES and FP7 EU Masterswitch grant; SG received the FP7 EU Masterswitch grant; and JHW received the Distler IZKF and DFG grant.
Competing interests JHWD has consultancy relationships and/or has received research funding from Boehringer Ingelheim, Celgene, Bayer Pharma, Actelion, Pfizer, Ergonex, BMS, JB Therapeutics, Anaphore, Inc, Sanofi-Aventis, Novartis, Array Biopharma and Active Biotec in the area of potential treatments of scleroderma and is stock owner of 4D Science. OD has consultancy relationship and/or has received research funding from Actelion, Pfizer, Ergonex, BMS, Sanofi-Aventis, United BioSource Corporation, medac in the area of potential treatments of scleroderma and its complications. He has received lecture honoraria from Actelion, Pfizer, Encysive and Ergonex. The real or perceived potential conflicts listed above are accurately stated. All others were supported by their respective institutions.
Provenance and peer review Not commissioned; externally peer reviewed.