Article Text

Abstract
Objectives Transforming growth factor β (TGFβ) has been identified as a key player in fibrotic diseases. However, the molecular mechanisms by which TGFβ activates fibroblasts are incompletely understood. Here, the role of JunD, a member of the activator protein 1 (AP-1) family of transcription factors, as a downstream mediator of TGFβ signalling in systemic sclerosis (SSc), was investigated.
Methods The expression of JunD was analysed by real-time PCR, immunofluorescence, western blotting and immunohistochemistry. The canonical Smad pathway was specifically targeted by small interfering (si)RNA. The expression of extracellular matrix proteins in JunD deficient (JunD−/−) fibroblasts was analysed by real-time PCR and hydroxyproline assays. The mouse model of bleomycin-induced dermal fibrosis was used to assess the role of JunD in experimental fibrosis.
Results JunD was overexpressed in SSc skin and in cultured fibroblasts in a TGFβ dependent manner. The expression of JunD colocalised with pSmad 3 in fibrotic skin and silencing of Smad 3 or Smad 4 by siRNA prevented the induction of JunD by TGFβ. JunD−/− fibroblasts were less responsive to TGFβ and released less collagen upon stimulation with TGFβ. Moreover, JunD−/− mice were protected from bleomycin-induced fibrosis with reduced dermal thickening, decreased myofibroblast counts and lower collagen content of lesional skin.
Conclusions These data demonstrate that JunD is overexpressed in SSc and that JunD is a mediator of the profibrotic effects of TGFβ. Considering that inhibitors of AP-1 signalling have recently been developed and are available for clinical trials in SSc, these findings may have translational implications.
Statistics from Altmetric.com
Introduction
Systemic sclerosis (SSc) is an autoimmune connective tissue disease that affects the skin and a variety of internal organs including the lungs, heart and gastrointestinal tract. The histopathological hallmarks of early stages of SSc are perivascular inflammatory infiltrates and a reduced capillary density.1,–,3 Later stages are dominated by an excessive accumulation of extracellular matrix components, which is caused by a pathological activation of fibroblasts.4 Transforming growth factor β (TGFβ) has been identified as a central mediator of fibroblast activation in SSc.4 However, the intracellular signalling cascades, by which this cytokine stimulates the production of extracellular matrix, are only partially understood, and current knowledge has not yet been translated into antifibrotic therapies for clinical use.5,–,7
JunD belongs to the Jun (c-Jun, JunB) family, which either forms homodimers or heterodimers with members of the Fos (c-Fos, FosB, Fos-related antigen 1 (Fra-1) and Fra-2) family to regulate the transcription of target genes (Jochum, #17655; Wagner, #17656).6,–,8 Jun and Fos proteins are also summarised as activator protein 1 (AP-1) transcription factors. The capability to activate or suppress gene expression depends on the composition of the AP-1 complex, because the different dimers vary significantly in their DNA binding affinity.9 Mice lacking JunD are viable and, apart from male sterility, develop normally.10 However, deficiency of JunD impairs stress responses. Thus, fibroblasts deficient for JunD are particular susceptible to stress-induced apoptosis.11 In addition, mice lacking JunD show increased and progressive kidney damage after subtotal nephrectomy12 and demonstrate increased cardiomyocyte apoptosis and mortality upon pressure overload.13 Consistent with a role of JunD in stress responses and cellular activation, JunD is prominently induced in hepatic stellate cells upon activation.14 The induction of JunD is more pronounced than for any other member of the Jun family suggesting that JunD is of particular importance for the activation of hepatic stellate cells. Considering the prominent role of hepatic stellate cells in liver fibrosis,15,–,17 we hypothesised that JunD might also regulate the activation of fibroblasts and contribute to tissue fibrosis in SSc.
Materials and methods
Patients and fibroblast cultures
Fibroblast cultures were obtained from lesional skin biopsies of 20 patients with SSc and 12 healthy volunteers matched for age and sex. All patients with SSc fulfilled the American College of Rheumatology (ACR) criteria for SSc. The median age of patients with SSc was 53 years (range 32–78 years) and their median disease duration was 6 years (range 0.5–13 years); all patients had diffuse cutaneous SSc and positive anti-nuclear antibodies; none of the patients were positive for anti-centromere antibodies, but 10 were positive for anti-topoisomerase 1 antibodies. None of the patients were treated with immunosuppressive or other potentially disease-modifying drugs at the time of biopsy. In all, 15 biopsies were taken from involved skin of the mid volar surface of the forearm (15±2 cm proximal to the ulna styloid). Five additional biopsies were obtained from the non-involved skin of the upper arm. All patients and controls signed a consent form approved by the local institutional review boards.
Fibroblast cultures were obtained from patients with SSc and healthy volunteers and from mice deficient for JunD (JunD−/−) and wild-type littermates (JunD+/+), and were cultured in Dulbecco's modified Eagle medium (DMEM)-Ham's F-12 containing 10% heat-inactivated fetal calf serum (FCS), 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 100 units/ml penicillin, 100 μg/ml streptomycin, 2 mM l-glutamine and 2.5 μg/ml amphotericin B (all from Invitrogen, Karlsruhe, Germany). In selected experiments, fibroblasts were stimulated with recombinant TGFβ in a concentration of 10 ng/ml, respectively (R&D Systems, Wiesbaden-Nordenstadt, Germany). This latter concentration represents the standard concentration used for the stimulation of dermal fibroblasts.18 19 Fibroblasts from passages 4–8 were used for the experiments.
Western blot analysis
For western blotting, 10 μg of protein in Laemmli sample loading buffer were separated on 10% SDS-polyacrylamide gel and electrotransferred onto poly(vinylideneflouride) (PVDF) (Roth, Karlsruhe, Germany) membranes. After blocking with 5% non-fat dry milk for 1 h, blots were incubated with specific polyclonal primary antibodies (1:400) diluted in 2% non-fat dry milk overnight at 4°C. For the detection of JunD, polyclonal antibodies against JunD (Santa Cruz Biotechnology, Heidelberg, Germany) were used. Signals were visualised with an enhanced chemiluminescence (ECL) (GE Healthcare, Buchinghemshire, UK) Plus Western Blotting Detection System and exposed to high-performance chemiluminescence film.
Immunofluorescence staining
Fibroblasts were incubated for 24 h with 0.1% DMEM/F12 and stimulated for 1, 3, 6, 9, 24 h with 10 ng/ml of TGFβ. The cultured fibroblasts were first washed with 1× phosphate buffered saline and then fixed with 4% methanol free paraformaldehyde at room temperature for 10 min. Fixed cells were permeabilised for 5 min at room temperature with 0.25% Triton X-100. Next, cells were incubated with 5% (v/v) horse serum blocking solution for 1 h. JunD was detected by incubation with polyclonal rabbit anti-human JunD antibodies over night at 4°C. Irrelevant isotype antibodies were used as controls. Polyclonal goat anti-rabbit antibodies labelled with fluorescent dye Alexa Fluor 488 (Invitrogen, Darmstadt, Germany) were used as secondary antibodies. In addition, cell nuclei were stained with 4',6-diamidino-2-phenylindole (DAPI) (Santa Cruz Biotechnology, Heidelberg, Germany). Images were captured at a 200-fold magnification.
Bleomycin-induced dermal fibrosis in JunD deficient mice
JunD−/− mice on a C57/Bl6 background have been described previously.10 Wild-type C57/Bl6 (JunD+/+) littermates were used as controls. Skin fibrosis was induced in 6-week-old mice by local injections of bleomycin every other day for 4 weeks20; 100 μl of bleomycin dissolved in 0.9% NaCl at a concentration of 0.5 mg/ml was administered by subcutaneous injections in defined areas of 1 cm2 at the upper back. Subcutaneous injections of 100 μl 0.9% NaCl were used as controls. One group of JunD−/− mice and one group of JunD+/+ mice were challenged with bleomycin, whereas the remaining two groups were injected with NaCl. After 4 weeks, the mice were killed to analyse the dermal thickness, hydroxyproline content and the number of myofibroblasts in lesional skin. A total of 20 JunD−/− mice and 16 JunD+/+ littermates were used for these analyses.
Evaluation of dermal thickness
Lesional skin areas were excised, fixed in 4% formalin for 6 h and embedded in paraffin. Sections of 5 μm were cut and stained with haematoxylin (Merck, Darmstadt, Germany) and eosin (Sigma-Aldrich, Steinheim, Germany) stain. The dermal thickness was measured at 100-fold magnification by measuring the distance between the epidermal-dermal junction and the dermal-subcutaneous fat junction at three sites from lesional skin of each mouse.19 The analysis was performed in a blinded manner by two independent examiners.
Hydroxyproline assay
The amount of collagen protein in skin samples was determined via hydroxyproline assay.21 After digestion of punch biopsies (diameter 3 mm) in 6 M hydrochloric acid for 3 h at 120°C, the pH of the samples was adjusted to 6 with 6 M sodium hydroxide. Afterwards, 0.06 M chloramine T was added to each sample and incubated for 20 min at room temperature. Next, 3.15 M perchloric acid and 20% p-dimethylaminobenzaldehyde were added and samples were incubated for additional 20 min at 60°C (all the above from Sigma-Aldrich, Steinheim, Germany). The absorbance was determined at 557 nm with a Spectra MAX 190 microplate spectrophotometer (Molecular Devices, Sunnyvale, CA, USA).
Immunohistochemistry
Formalin-fixed, paraffin-embedded skin sections were stained with anti-JunD antibodies (Santa Cruz Biotechnology, Heidelberg, Germany) or anti-smooth muscle actin (SMA) (clone 1A4; Sigma-Aldrich, Steinheim, Germany) antibodies. Peroxidase labelled species-specific Igs (Dako, Glostrup, Denmark) were used as secondary antibodies. Irrelevant isotype matched antibodies were used as controls. Staining was visualised with 3,3'-diaminodbenzidine (DAB) (Sigma-Aldrich, Steinheim, Germany), peroxidase substrate solution (Sigma-Aldrich). αSMA stained sections were counterstained with haematoxylin. The number of myofibroblasts was determined at 200-fold magnification. Counting was performed in a blinded manner by two independent examiners.
Quantitative real time-PCR
Gene expression was quantified by SYBR Green real time-PCR using the ABI Prism 7300 Sequence Detection System (Applied Biosystems, Foster City, California, USA) as described.22 The following primer pairs were used: human JunD: 5‘-AAGAGTCAGAACACGGAGCTG-3’ (forward), 5‘-GGCTGAGGACTTTCTGCTTG-3’ (reverse); human Smad 3: 5‘-GCTGGGCTGAAGCGCACTGA-3’ (forward), 5‘-GGCTGCGAGGCGTGGAATGT-3’ (reverse); human Smad 4: 5‘-GCCACCCAAACCGCTCCGT-3’ (forward), 5‘-CACGGCCCTGGTCGTCGTC-3’ (reverse); murine JunD: 5‘-ACTACCCCGACCAGTACGC-3’ (forward), 5‘-TCGCTAGCTGCCACCTTC-3’ (reverse); murine col1a1: 5‘-GAAGCACGTCTGGTTTGGA-3’ (forward), 5‘-ACTCGAACGGGAATCCATC-3’ (reverse); murine col1a2: 5‘-TCAAACTGGCTGCCACCAT-3’ (forward), 5‘-CCAACAAGCATGTCTGGTTAGGA-3’ (reverse); murine plasminogen activator inhibitor 1 (PAI-1): 5‘-ACGTTGTGGAACTGCCCTAC-3’ (forward), 5‘-AGCGATGAACATGCTGAGG-3’ (reverse); murine Smad 7: 5‘-GCTCAATTCGGACAACAAGAG-3’ (forward), 5‘-TCTTGCTCCGCACTTTCTG-3’ (reverse); samples without enzyme in the reverse transcription reaction (non-RT controls) were used as negative controls. Unspecific signals caused by primer dimers were excluded by no template controls and by dissociation curve analysis. β-Actin was used to normalise for the amounts of cDNA within each sample.
Statistics
Data were expressed as mean±SEM. The Student t test was used for statistical analyses. A p value of less than 0.05 was considered statistically significant.
Results
JunD is overexpressed in patients with SSc
To analyse the expression of JunD in patients with SSc, we first determined the mRNA levels of JunD in skin biopsies from patients with SSc and healthy volunteers. The mRNA levels of JunD were increased by 3.1±0.3-fold in clinically involved skin of patients with SSc compared to controls (p=0.04) (figure 1A). A comparable upregulation of JunD mRNA was observed in clinically non-involved skin (3.1±0.3-fold). An upregulation of JunD in patients with SSc was also observed by immunohistochemistry of the dermis. A widespread expression of JunD was observed in skin sections from clinically involved skin of patients with SSc with a prominent staining in fibroblasts and endothelial cells, but also in keratinocytes (figure 1B). The expression pattern in clinically non-involved skin was identical to that in involved skin. In contrast to patients with SSc, the expression of JunD was largely restricted to keratinocytes in healthy individuals and either absent or very weak in fibroblasts and endothelial cells.
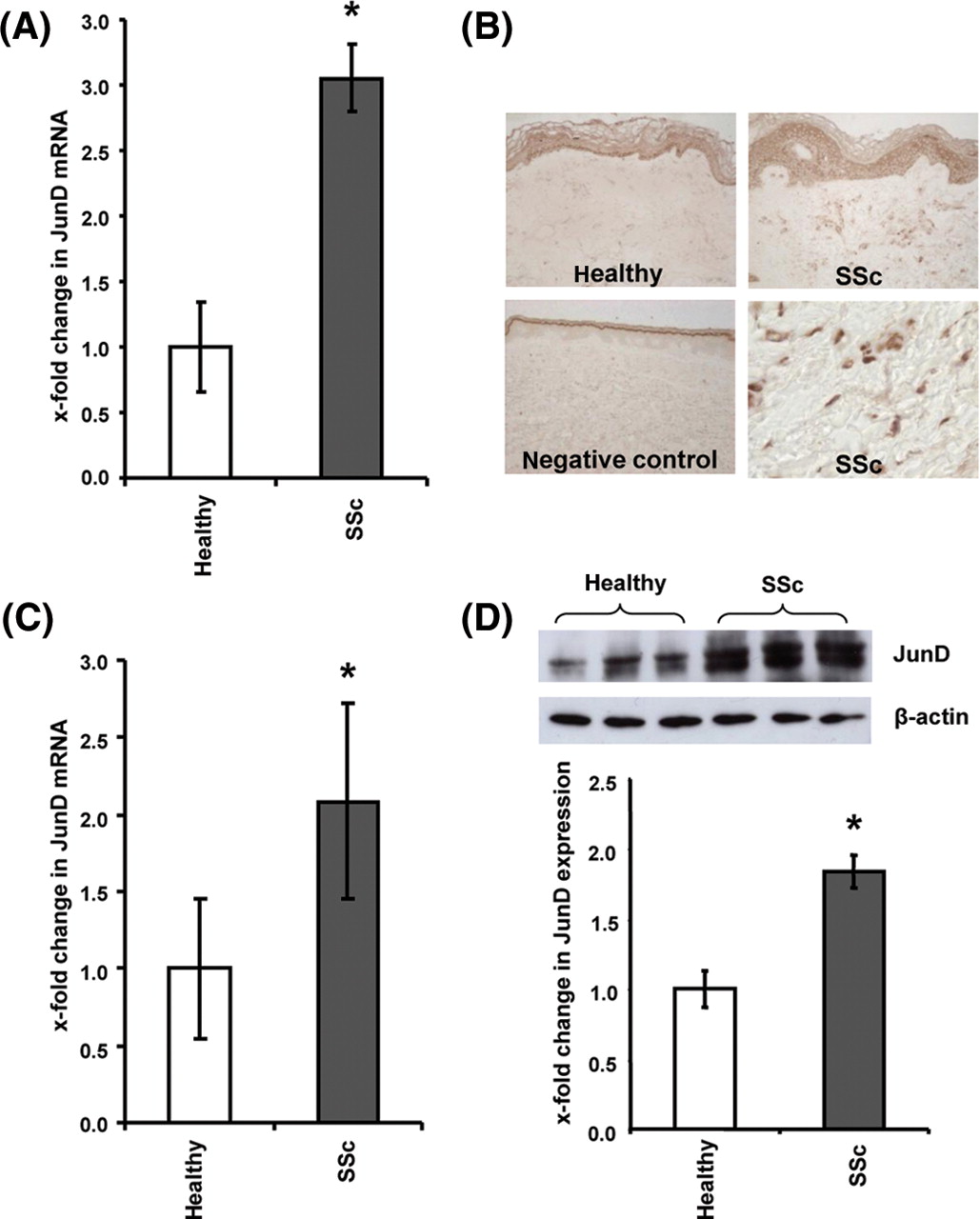
JunD is overexpressed in patients with systemic sclerosis (SSc). (A) The mRNA levels of JunD were elevated in the skin of patients with SSc compared to healthy controls. (B) A prominent staining for JunD protein was observed by immunohistochemistry in fibroblasts, keratinocytes and endothelial cells of patients with SSc. In contrast, the expression of JunD was significantly reduced in age-matched and sex-matched healthy individuals (n=7 for both). Two representative overview sections and a negative (isotype) control are shown at 200-fold magnification. In addition, a staining from a patient with SSc is shown at 1000-fold magnification to show the intense staining in fibroblasts and vessels. The epidermis is marked with E, the dermis with D. Some representative fibroblasts are marked with arrows, vessels with arrowheads. (C and D) The overexpression of JunD in fibroblasts persisted in vitro. (C) Increased mRNA and (D) protein levels of JunD were detected by real-time PCR and western blot in SSc fibroblasts compared to control cells. *Indicates statistical significant differences as compared to healthy volunteers and control fibroblasts. F, spindle shaped fibroblasts; V, vessels
Increased levels of JunD were also observed in cultured fibroblasts of patients with SSc. The mRNA levels of JunD were significantly increased in SSc by 2.2±0.3-fold compared to control fibroblasts (p=0.03) (figure 1C). Consistently, the protein levels of JunD were also upregulated in SSc fibroblasts by 1.8±0.1-fold (p=0.01) (figure 1D).
TGFβ stimulates the expression of JunD in a Smad dependent manner
We hypothesised that the upregulation of JunD in SSc fibroblasts might be mediated by TGFβ. Indeed, incubation of fibroblasts with TGFβ stimulated the expression of JunD and induced nuclear accumulation of JunD. The expression of JunD mRNA and protein increased within 1 h after stimulation with TGFβ and the protein levels remained elevated for 24 h (figure 2A,B). JunD was located in the cytoplasm in resting cells and within the first 3 h after stimulation, but prominently accumulated in the nucleus between 6 and 9 h after stimulation with TGFβ (figure 2B).

Transforming growth factor (TGF)β induces JunD in a Smad-dependent manner. (A) Stimulation with TGFβ induced the mRNA levels of JunD in healthy dermal fibroblasts in a time-dependent manner with a maximal induction after 1 h. (B) TGFβ increased the protein levels of JunD and induced nuclear translocation in healthy dermal fibroblasts as analysed by immunofluorescence. The expression of JunD was increased after stimulation with TGFβ for 1 h and translocated into the nuclei after 6 h. The expression of JunD protein was visualised with fluorescein isothiocyanate (FITC)-labelled secondary antibodies (green staining) and nuclei of fibroblasts were stained with 4',6-diamidino-2-phenylindole (DAPI) (blue staining). (C) Transfection of fibroblasts from healthy volunteers with small interfering (si)RNA against Smad 3 prevented the induction of JunD by TGFβ. (D) Phosphorylated and thereby activated Smad 3 (red staining) and JunD (brown staining) colocalise in skin biopsies of patients with systemic sclerosis (SSc) as detected by immunohistochemistry. Representative tissue sections are shown at 200-fold (upper images), a detailed view of JunD and pSmad 3 double positive stained cells is shown in SSc sections at 1000-fold magnification (lower images). *Indicates statistical significant differences as compared to unstimulated fibroblasts (A and B) or compared to fibroblasts transfected with non-targeting siRNA (C).
To determine whether TGFβ induces JunD via the canonical Smad pathway, the expression of Smad 3 and Smad 4 was targeted by small interfering (si)RNA in human fibroblasts. siRNA against Smad 3 and Smad 4 reduced the mRNA and protein levels of the respective proteins by >85% (data not shown). Knockdown of Smad 3 almost completely prevented the induction of JunD by TGFβ with a decrease of 80±14% (p=0.04) compared to fibroblasts transfected with mock siRNA (figure 2C). siRNA against Smad 4 also prevented the upregulation of JunD by TGFβ (data not shown), demonstrating that the canonical Smad cascade is essential for the induction of JunD by TGFβ.
To confirm the central roles of TGFβ and Smad signalling for the induction of JunD in SSc, SSc sections were stained for phosphorylated and thus activated Smad 3 (pSmad 3) and JunD. Double staining revealed colocalisation of JunD with pSmad 3 in >90% of JunD positive cells. Vice versa, virtually all cells positive for pSmad 3 expressed high levels of JunD (figure 2D).
Lack of JunD reduces the stimulatory effects of TGFβ on collagen synthesis
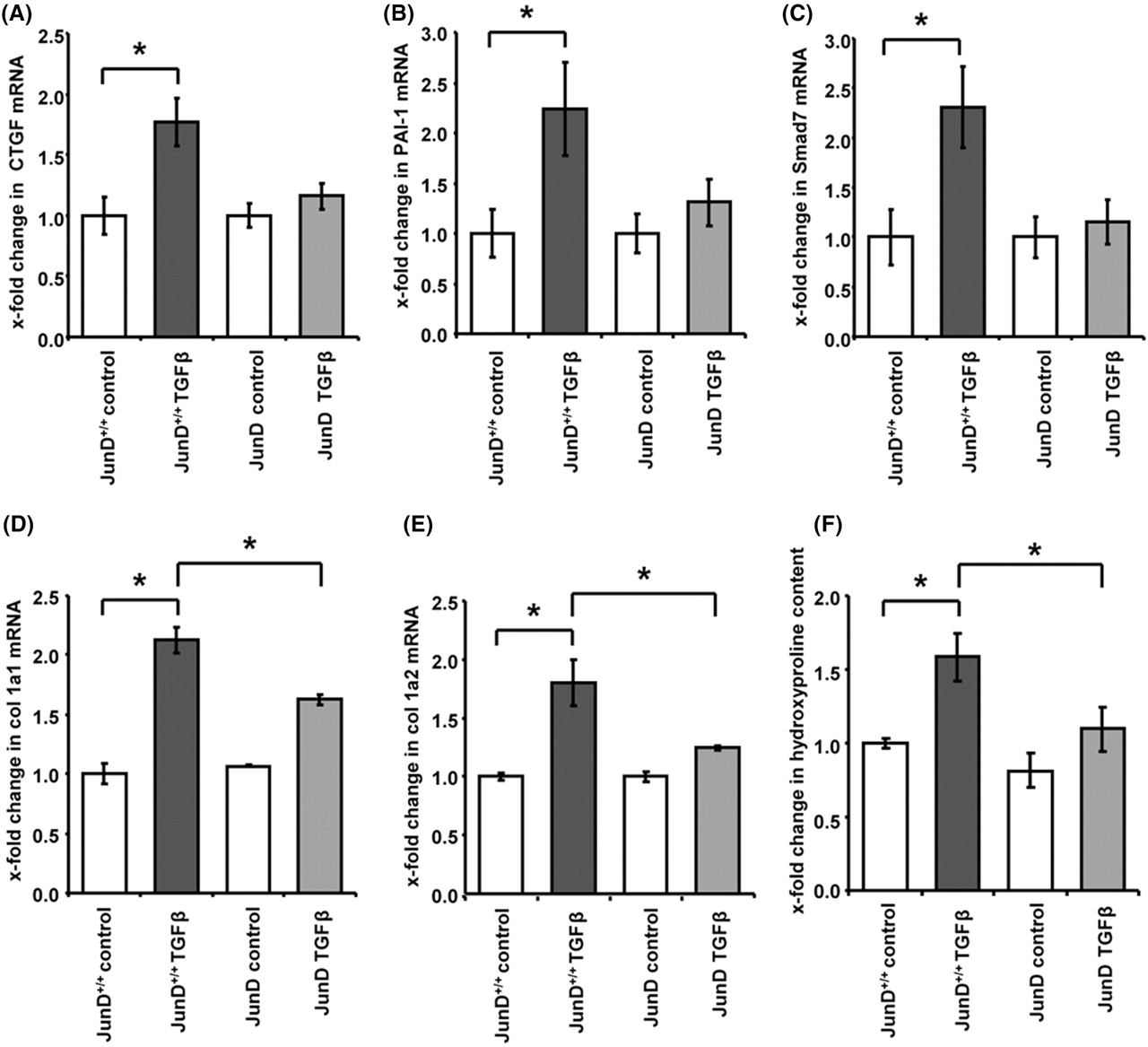
We next investigated, whether the induction of JunD might contribute to the profibrotic effects of TGFβ. We first analysed, whether deficiency of JunD might decrease the expression of classical TGFβ target genes in fibroblasts. Indeed, the stimulatory effects of TGFβ on the mRNA levels of connective tissue growth factor (CTGF) were decreased by 80±11% in JunD−/− fibroblasts compared to JunD+/+ cells (p=0.02) (figure 3A). The induction of PAI-1 and Smad 7 were also decreased by 75±23% and 88±22% in JunD−/− fibroblasts (p=0.01 for PAI-1 and p=0.04 for Smad 7) (figure 3B,C) demonstrating a decreased responsiveness of JunD−/− fibroblasts towards TGFβ.

Deficiency of JunD reduces the responsiveness towards transforming growth factor (TGF)β. (A–C) The induction of typical TGFβ target genes such as (A) connective tissue growth factor (CTGF), (B) plasminogen activator inhibitor 1 (PAI-1) and (C) Smad 7 was reduced in fibroblasts deficient for JunD compared to controls. (D–F) The stimulatory effects of TGFβ on the mRNA levels of (D) col 1a1, (E) col 1a2 as well as the increased release of (F) hydroxyproline were also decreased in JunD−/− fibroblasts compared to controls. *Indicates statistical significant differences as compared to wild-type cells.
The upregulation of type I collagens upon stimulation with TGFβ was also reduced in JunD−/− fibroblasts. The induction of collagen type 1, alpha 1 (col 1a1) and col 1a2 were decreased by 46±15% and 70±7% in JunD−/− fibroblasts compared to JunD+/+ fibroblasts (p=0.04 for col 1a1 and p=0.03 for col 1a2) (figure 3D,E). Similar findings were also obtained on the protein level with a significant reduction of the TGFβ-induced hydroxyproline content in JunD−/− by 83±15% (p=0.03) (figure 3F). Consistent with the low expression of JunD in resting fibroblasts, the basal collagen synthesis was not affected in JunD−/− fibroblasts.
Mice deficient for JunD have ameliorated bleomycin-induced dermal fibrosis
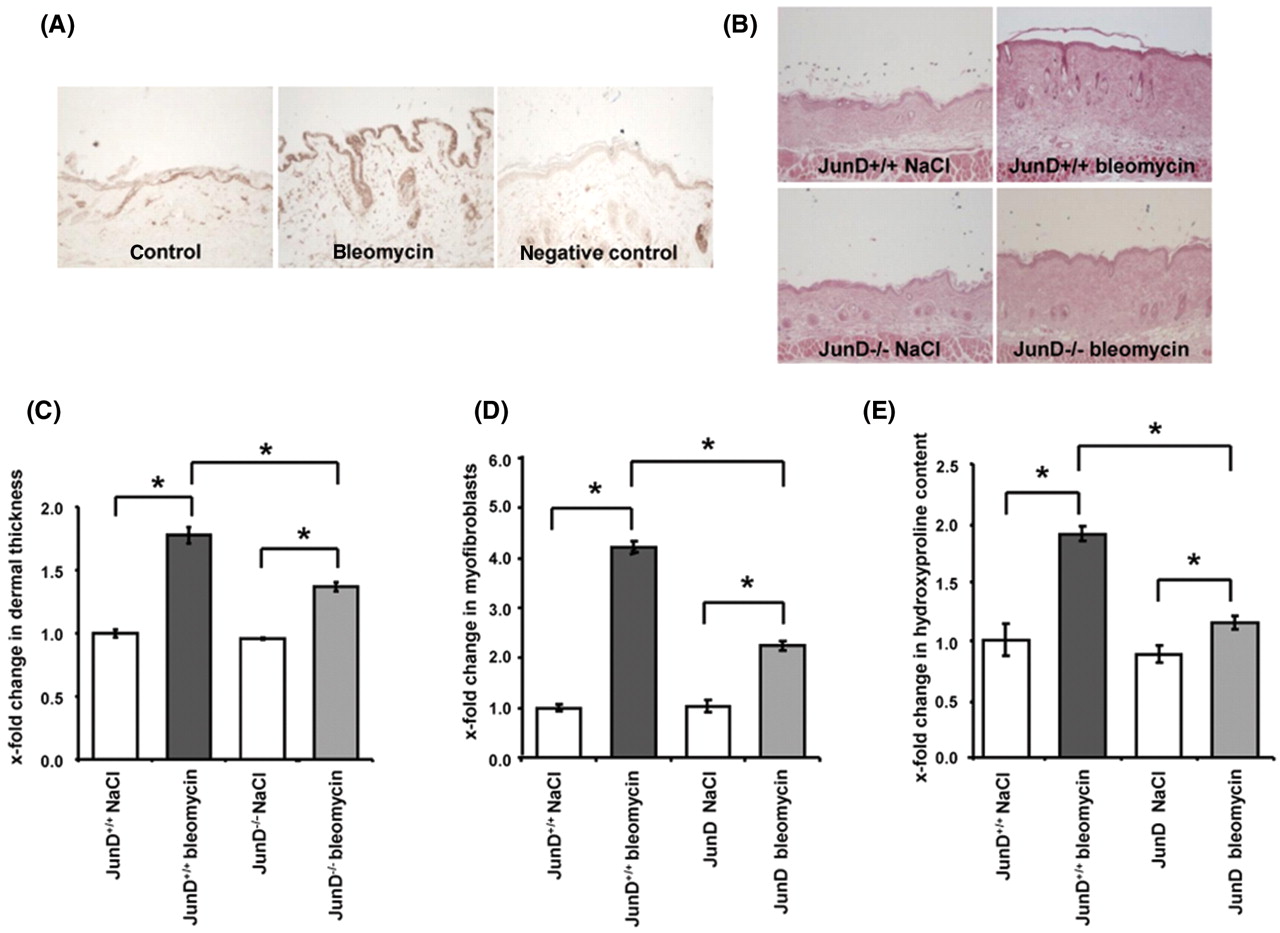
To study the role of JunD in experimental fibrosis, the mouse model of bleomycin-induced dermal fibrosis was used. Similar to the findings in patients with SSc, an increased expression of JunD was also observed in wild-type mice challenged with bleomycin (figure 4A), confirming that the model of bleomycin-induced fibrosis is suitable to study the role of JunD.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
JunD is crucial for the development of experimental fibrosis. (A) The expression of JunD was increased in the mouse model of bleomycin-induced skin fibrosis. Representative images of control mice injected with NaCl, of mice challenged with bleomycin and of a control section incubated with isotype control antibodies are shown at 200-fold magnification. (B) Reduced dermal thickening in mice lacking JunD. Representative tissue sections at 100-fold magnification are shown: JunD+/+ mice injected with NaCl intracutaneously (n=7), JunD−/− mice with intracutaneous injections of NaCl (n=8), bleomycin-injected JunD+/+ mice (n=7), JunD−/− mice injected with bleomycin (n=6). (C) Deficiency of JunD prevented the differentiation of resting fibroblasts into myofibroblasts upon challenge with bleomycin. (D) Decreased collagen content in lesional skin of bleomycin-challenged JunD−/− mice compared to wild-type littermates. In total, 20 JunD−/− mice and 16 JunD +/+ littermates were analysed. *Indicates statistical significant differences compared to bleomycin-injected JunD+/+ mice.
No differences in skin histology were observed between JunD−/− mice and JunD+/+ mice in the absence of a fibrotic stimulus (figure 4B). Injections of bleomycin-induced prominent fibrosis in JunD+/+ with increased dermal thickness, activation of fibroblasts and accumulation of collagen (figure 4C–E). The fibrotic effects to bleomycin were significantly ameliorated in JunD−/− mice. Dermal thickening in lesional skin was reduced by 53±8% in JunD−/− mice compared to JunD+/+ mice (p=0.002) (figure 4C). The number of myofibroblasts and the hydroxyproline content in lesional skin were also significantly reduced by 64±9% and 84±6%, respectively (p=0.0001 for the number of myofibroblasts and p=0.003 for the hydroxyproline content) (figure 4D,E). All outcomes remained statistically significant after Bonferroni correction for multiple testing.
Discussion
The present study demonstrates a role of JunD as a downstream mediator of the profibrotic effects of TGFβ in SSc fibroblasts. (1) JunD is induced by TGFβ within 1 h and its expression colocalises with phosphorylated Smad 3 as a marker for active TGFβ signalling in vivo. (2) The expression of classical TGFβ target genes such as PAI-1, Smad 7 and CTGF is prevented in fibroblasts deficient for JunD. (3) Deficiency for JunD abrogates the TGFβ-induced differentiation of resting fibroblasts into myofibroblasts. (4) The stimulatory effects of TGFβ on collagen synthesis are reduced in fibroblasts lacking JunD. (5) JunD also regulates the expression of other profibrotic mediators such as interleukin 6 and tissue inhibitor of matrix metalloproteinases 1 (TIMP-1).23,–,25 (6) JunD−/− mice are protected from bleomycin-induced experimental fibrosis. These findings may have direct translational implications. The mild phenotype of mice completely deficient for JunD10 suggests that pharmacological targeting of JunD may not be prevented by toxicity. Although specific inhibitors for JunD are currently not available, small molecule inhibitors of AP-1 signalling have recently been described and are currently tested in clinical trials.26 However, further studies are needed to evaluate JunD as a potential target for antifibrotic therapies. In particular, the role of JunD for fibrosis should be confirmed in additional, less inflammatory mouse models of SSc.
We demonstrated that JunD is overexpressed in fibrotic skin of patients with SSc and that this overexpression is induced by TGFβ via activation of canonical Smad signalling. Increased levels of JunD were also detected in cultured SSc fibroblasts, consistent with the characteristic autocrine activation of TGFβ in SSc fibroblasts in vitro.27 28 Considering the role of JunD for the stimulatory effects of TGFβ on fibroblast activation, the upregulation of JunD in all SSc lines suggests that the persistent activation of JunD in SSc fibroblasts might contribute to the characteristic activated phenotype of SSc fibroblasts with persistently increased expression of myofibroblast markers and collagen synthesis even in the absence of exogenous stimuli and after several passages in culture. Interestingly, we did not observe differences in the expression levels of JunD between patients positive and negative for anti-topoisomerase or anti-centromere autoantibodies, suggesting that the status of conventional autoantibodies is not related to the expression of JunD.
Although it was beyond the scope of our study, JunD might also contribute to vascular disease in SSc by functioning as a dimerisation partner of Fra-2. Fra-2 is overexpressed in vessels of patients with SSc and transgenic overexpression of Fra-2 in mice results in proliferation of pulmonary vascular smooth muscle cells and in histological changes resembling those of pulmonary arterial hypertension in humans, and also in a progressive microangiopathy with capillary rarefication.29 30 Fra-2 dimerises with members of the Jun family of AP-1 transcription factors to regulate the expression of target genes. JunD seems to be the preferred dimerisation partner in the context of TGFβ signalling as evidenced by studies on the laminin α3 gene.31 The hypothesis of a role of JunD/Fra-2 dimers in SSc vasculopathy is further supported by the expression patterns of both genes in patients with SSc. The expression pattern of JunD was identical to that of Fra-2 and included a prominent expression of JunD and Fra-2 in endothelial cells.32 However, further studies are needed to address a potential role of JunD in the vascular manifestations of SSc.
In summary, we identify JunD as mediator of TGFβ signalling in SSc. JunD is overexpressed in a TGFβ/Smad dependent manner in SSc and mediates the profibrotic effects of TGFβ on SSc fibroblasts. Deficiency of JunD reduces the activation of fibroblasts and prevents experimental fibrosis. These findings might have translational implications, because inhibitors of AP-1 signalling have been developed and would be available for clinical trials in SSc. However, further studies are needed to evaluate JunD as a potential therapeutic target in SSc.
Acknowledgments
We thank Maria Halter and Anna-Maria Herrmann for excellent technical assistance.
References
Footnotes
KP and PZ contributed equally to this work.
-
Funding Grant A40 of the Interdisciplinary Center of Clinical Research (IZKF) in Erlangen, grants from the Deutsche Forschungsgesellschaft, CMH Research Projects No 00000023728 and the Career Support Award of Medicine of the Ernst Jung Foundation (to JHWD).
-
Competing interests None.
-
Ethics approval This study was conducted with the approval of the University of Erlangen-Nuremberg.
-
Provenance and peer review Not commissioned; externally peer reviewed.