Article Text

Abstract
Objective Serum amyloid A (A-SAA) is an acute-phase protein with cytokine-like properties implicated in the pathogenesis of rheumatoid arthritis (RA), atherosclerosis, diabetes and Alzheimer's disease. This study characterises the mechanism of A-SAA-induced cytoskeletal rearrangement and migration in synovial fibroblasts and microvascular endothelial cells (human dermal endothelial cells; HDEC).
Methods Immunohistology and immunofluorescence were used to examine αvβ3 and β1-integrins, filamentous actin (F-actin) and focal adhesion expression in rheumatoid arthritis synovial tissue (RAST) and rheumatoid arthritis synovial fibroblast cells (RASFC). A-SAA-induced αvβ3 and β1-integrin binding was measured by adhesion assay. Cytoskeletal rearrangement and ρ-GTPase activation following A-SAA stimulation was examined using dual immunofluorescent staining for F-actin/vinculin staining, pull down assays and immunoblotting for Cdc42 and RhoA. Cell growth, invasion/migration, angiogenesis and actin formation were examined in the presence or absence of specific Rac1 and Cdc42 inhibitors (NSC23766 and 187-1).
Results αvβ3, β1-integrin and F-actin predominantly localised to vascular endothelium and lining layer cells in RAST, compared with osteoarthritis and normal control synovial tissue. A-SAA significantly increased αvβ3 and β1 binding in RASFC. A-SAA induced cytoskeletal disassembly, loss of focal adhesions and filopodia formation in RASFC and HDEC. A-SAA significantly induced Cdc42 activation but failed to promote RhoA activation in HDEC and synovial fibroblast cells. Blockade of Rac-1 and Cdc42 inhibited A-SAA-induced cell growth, invasion/migration, actin cytoskeletal rearrangement and angiogenesis.
Conclusions These data show a novel mechanism for A-SAA-induced cell migrational events in RA mediated via cytoskeletal signalling pathways.
Statistics from Altmetric.com
Rheumatoid arthritis (RA) is a progressive autoimmune disease. One early inflammatory event is angiogenesis in which new blood vessels invade synovial tissue facilitating a self-perpetuating infiltration of immune cells. This results in lining layer hyperplasia and the destruction of adjacent articular cartilage. Serum amyloid A (A-SAA) is an acute-phase protein of the apoprotein family synthesised primarily by hepatocytes;1 2 however, extrahepatic production has been described in RA, Alzheimer's and cardiovascular disease.3,–,6 Increased expression of A-SAA and its receptors, scavenger receptor class B1, formyl peptide receptor-like 1 (FPRL-1) and Toll-like receptor 2 (TLR2), in rheumatoid arthritis synovial tissue (RAST).5 7 8 A-SAA induces angiogenesis, adhesion, leucocyte recruitment, chemokine expression and metalloproteinases in RA cells in vitro.9,–,11 In a human RA/severe combined immunodeficient mouse chimera model we demonstrated that A-SAA induces cell migration and angiogenesis in vivo.10 Endogenous A-SAA from RA plasma has little effect on neutrophil shedding;12 however, studies have demonstrated a functional role for endogenous A-SAA in RA cells.10 13
Cell migration and altered vascular morphology are fundamental processes in RAST infiltration and invasion. When activated immune cells migrate, they become polarised forming membrane protrusions at the leading edge.14 This process depends on integrin–focal adhesion kinase (FAK) signalling pathways. Integrins activated on the cell membrane form clusters of focal adhesion contacts that bind to extracellular matrix (ECM). This process is accompanied by the recruitment of cytoskeletal proteins including paxillin, vinculin and actin, important scaffold molecules to regulate cytoskeletal assembly/disassembly. Engagement of integrin receptors results in the downstream activation of ρ-family GTPase proteins, Rac-1, RhoA and Cdc42, involved in actin/stress fibre maintenance and turnover. This stimulates cell contraction, which allows cell body movement to form filopodia, lamellipodia and membrane ruffling. In blood vessel morphogenesis ρ-GTPase activation is important for endothelial cell branching/sprouting and vacuolation in addition to lumen formation.15,–,19 Integrins are implicated in RA pathogenesis including angiogenesis and cartilage invasion. Rheumatoid arthritis synovial fibroblast cell (RASFC) β1-integrin binds to ECM components, an essential step in cartilage invasion. FAK and paxillin are expressed in RAST, macrophages and RASFC,20 21 whereas the inhibition of ρ-GTPase and FAK pathways inhibit RASFC adhesion, spreading, migration and invasiveness, demonstrating key roles in these processes.
Whereas A-SAA appears critical for RA cell migration and angiogenesis, little is known about the signalling pathways involved. We examined whether A-SAA induces reorganisation of the actin cytoskeleton through the activation of ρ-GTPases, to induce morphological changes in RASFC and endothelial cells.
Methods
RA patients with actively inflamed knee joints, classified using the American College of Rheumatology criteria,22 osteoarthritis and healthy controls were recruited from St Vincent's University Hospital outpatients. This research was performed in accordance with the Declaration of Helsinki, approved by the St Vincent's University Hospital ethics committee, following fully informed written consent. Synovial tissue biopsies were obtained by arthroscopy under local anaesthetic, using a 2.7 mm telescope (Stortz, Tuttlingen, Germany) as previously described.23 Biopsies were collagenase-digested to isolate primary RASFC or snap frozen in OCT and stored at −80°C.
RASFC isolation and culture
Synovial tissue was digested with 1 mg/ml collagenase type 1 (Worthington Biochemical, Dublin, New Jersey, USA) in RPMI (Gibco-BRL, Paisley, UK) for 4 h at 37°C in humidified air with 5% carbon dioxide. Dissociated cells grown to confluence in RPMI 1640, 10% fetal calf serum (Gibco-BRL), 10 ml 1 mmol/l HEPES (Gibco-BRL), penicillin (100 units/ml; Biosciences, Oxford, UK), streptomycin (100 units/ml; Biosciences) and fungizone (0.25 μg/ml; Biosciences) were used between passages 4 and 8. K4-immortalised synoviocyte fibroblast cell (SFC) line (kind gift of Dr Evelyn Murphy), were also cultured and used between passages 35 and 38. RASFC and K4 were rendered quiescent by omitting serum for 24 h, then stimulated with A-SAA (10, 50 µg/ml; Peprotech, Rocky Hill, NJ) or tumour necrosis factor alpha (TNFα) (10 ng/ml; R&D Systems, Abingdon, UK). Endotoxin levels of 1 EU or less in A-SAA preparations were confirmed by Limulus assay (BioWhittaker, Walkersville, MD).
Endothelial cells
Human dermal endothelial cells (HDEC; Lonza Wokingham, Berkshire, UK) were incubated in endothelial basal medium supplemented with endothelial growth medium-microvascular bullet kit containing 25 ml fetal calf serum, 0.5 ml human epidermal growth factor, 0.5 ml hydrocortisone, 0.5 ml gentamicin and 0.5 ml bovine brain extract. Serum (1%) was added 24 h before stimulation with A-SAA (10, 50 μg/ml) or TNFα (10 ng/ml).
Immunohistochemistry
Sections (7 μm) were cryostat cut, fixed in acetone for 10 min and air-dried. Non-specific binding and endogenous peroxidase activity was blocked using 10% casein and 0.3% hydrogen peroxide, respectively. A routine three-stage immunoperoxidase technique incorporating avidin-biotin–immunoperoxidase complex (DAKO, Dublin, Ireland) was used. Separate sections were incubated with primary mouse monoclonal antibodies to anti-αvβ3 and anti-βI (R&D Systems) at room temperature for 1 h. A secondary rabbit/mouse horseradish peroxidise (HRP) solution was added for 30 min. Colour was developed with diaminobenzidine-tetrahydrochloride (Sigma, St Louis, Missouri, USA), counterstained with haematoxylin and mounted. For filamentous actin (F-actin), slides were stained with FITC-phalloidin (Molecular Probes, Eugene, OR) for 40 min at room temperature, washed then counterstained with 4′,6-diamidino-2-phenylindole nuclear stain for 5 min and mounted in antifade fluorescent medium. An isotype-matched non-immune IgG was the negative control for all experiments. Sections were assessed using light microscopy.
Immunofluorescence
RASFC and HDEC were seeded into eight-well chamber slides, serum starved for 24 h before A-SAA (10 μg/ml) stimulation. Slides were rinsed in phosphate-buffered saline (PBS), fixed in 3.7% paraformaldehyde for 20 min and permeabilised with 0.1% triton X-100. Slides were blocked for 1 h in 5% bovine serum albumin before incubation with vinculin primary monoclonal antibodies (Sigma) at 4°C overnight, then rinsed and incubated with FITC-conjugated secondary antibody (Invitrogen, Dublin, Ireland) for 1 h in the dark. F-actin is visualised with FITC-phalloidin-stained slides for 40 min at room temperature, p-paxillin, by incubating for 1 h with fluorescent secondary antibody Cy3. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole. Stained cells were visualised with a Leitz DM40 microscope (Leica Microsystems, Wetzlar, Germany). For cytoskeletal inhibition experiments, cells were serum starved for 24 h in the presence/absence of the Rac1 inhibitor NCS23766 (50 µM), Cdc42 inhibitor 187-1 (10 µM), RhoA inhibitor Y27632 (10 µM; Calbiochem, Merck Chemicals, Nottingham, UK) and 1% dimethyl sulfoxide (DMSO) vehicle control before 24 h stimulation with A-SAA.
Integrin assay
Cell surface integrin binding to anti-integrin-coated plates was quantified using immunoadhesion assays (Chemicon, Temecula, California, USA). Ninety-six well trays are coated with anti-αvβ3 integrin or anti-β1 integrin mouse monoclonal antibodies. Goat antimouse IgG-treated wells blocked with irrelevant mouse IgG are negative controls. RASFC (5×104) seeded onto the coated wells were incubated for 1 h at 37°C with A-SAA (10 μg/ml). Following three washes, adherent cells were fixed, stained with crystal violet and quantified by measuring absorbance at 560 nm.
Pull down assays
Cdc42 and RhoA activity was determined using EZ-Detect Activation Kits (Pierce, Rockford, Illinois, USA) as per the manufacturer's protocol. SFC and HDEC were stimulated for 6 and 12 h time periods with A-SAA (10 μg/ml) or TNFα (10 ng/ml). After treatment, cells were rinsed once with ice cold PBS and scraped into 500 μl of lysis buffer. Samples were vortexed, centrifuged at 1600g for 15 min at 4°C before the supernatant was transferred to a new tube. Active Cdc42 and RhoA were affinity purified with GST-Pak1-PBD (Pak 1-p21-binding domain) and GST-Rhotekin, separated by sodium dodecylsulphate polyacrylamide gel electrophoresis and measured by western blotting using anti-Cdc42 and anti-ρ antibodies. Blots were developed using ECL (Pierce) for the detection of HRP and normalised to β-actin expression following densitometric analysis of detected protein bands.
Cell growth assay
RASFC were grown to approximately 70% confluence on 24-well plates before serum starving in RPMI-1640 for 24 h, followed by incubation with A-SAA (10 µg/ml) with or without the Rac1 inhibitor NCS23766 (50 µM), Cdc42 inhibitor 187-1 (10 µM) or 1% DMSO control in duplicate wells for 24 h. Cells were fixed for 15 min in 1% glutaraldehyde, PBS washed, then to supernatants 1 ml of 0.2% crystal violet solution was added to each well for 10 min. Stained cells were fully solubilised using 400 μl 1% sodium dodecylsulphate, 100 μl of this solution was transferred into a separate 96-well plate and absorbance read at 530 nm.
Transwell invasion assay
HDEC (2.5×104) were seeded in Biocoat Matrigel Invasion Chambers (Becton Dickinson, Oxford, UK) on 8 µm membranes containing A-SAA (10–50 µg/ml). Lower wells were incubated with anti-β1 integrin neutralising antibody, an appropriate IgG control or NCS23766, 187-1 (10 µM) or 1% DMSO control. Cells were allowed to migrate for 24 h in 1% endothelial basal medium. Migrating cells attached to the lower membrane were fixed with 1% glutaraldehyde and stained with 0.1% crystal violet. To assess the average number of migrating cells, five random high power fields were counted.
Wound repair assay
RASFC were seeded in 48-well plates and grown to confluency, growth arrested for 24 h in serum-free RPMI and a sterile pipette tip was used to create a linear wound across the well. HDEC and RASFC were stimulated with A-SAA (10 μg/ml) in the presence or absence of anti-β1 integrin neutralising antibody and an appropriate IgG control or NCS23766 (50 µM), 187-1 (10 µM) or 1% DMSO control for 24 h. Migration was determined by visual assessment of cells migrating into the wound using phase contrast microscopy.
HDEC tubule formation in vitro
Matrigel (50 μl; Becton Dickinson, Mountain View, California, USA) was plated in 96-well culture plates and allowed to polymerise for 1 h, at 37°C in humidified air with 5% carbon dioxide; 250 μl of HDEC cell suspension (4×104 cells/ml) was added to each well in the presence of A-SAA (10 μg/ml), with or without NCS23766 (50 µM), 187-1 (10 µM) or 1% DMSO control for 24 h. Tubule formation was assessed and photographed under phase-contrast microscopy as previously described.24
Statistics
SPSS15 was used for statistical analysis. The non-parametric Wilcoxon signed rank test and parametric Student's t tests were used for analysis; p<0.05 was considered statistically significant.
Results
Integrin and F-actin expression
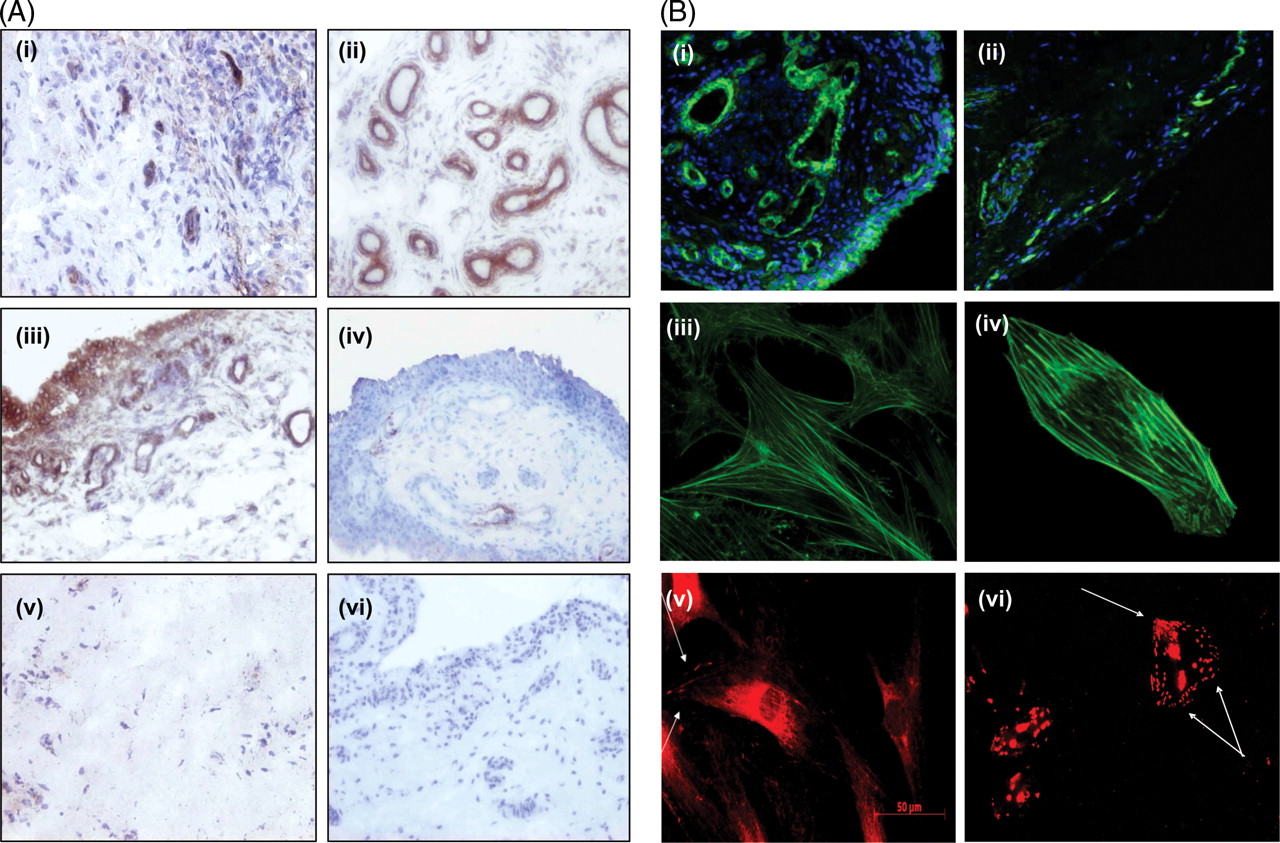
RAST αvβ3 expression was localised to endothelium with some expression observed in the lining layer (figure 1A(i)). β1-integrin was strongly expressed in RAST localised to both the endothelium and lining layer consistent with previous reports (figure 1A(ii), (iii)).21 25 αvβ3 and β1 expression was minimally expressed in osteoarthritis or normal synovial tissue. Representative images of αvβ3 in osteoarthritis (iv) and β1 in normal synovium (v) are demonstrated.
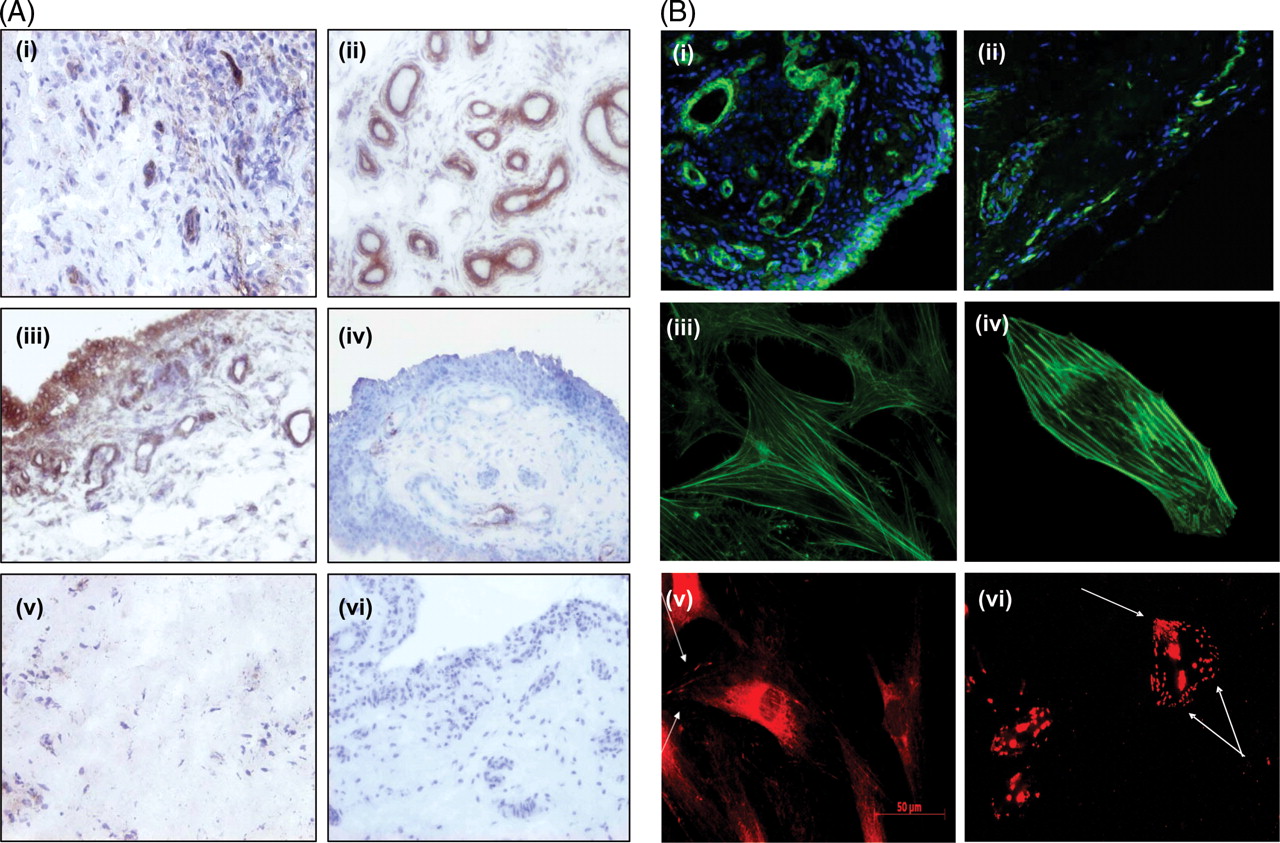
Expression of αvβ3, β1 and filamentous actin (F-actin) in rheumatoid arthritis synovial tissue (RAST). (A) Representative images of αvβ3 (i) and β1 (ii) in RAST. β1 integrin was strongly expressed in rheumatoid arthritis (RA) synovium localised to both the vascular endothelium (ii) and the lining layer regions (iii). Representative image of αvβ3 in osteoarthritis (iv), β1 integrin in normal synovial tissue (v) and the IgG isotype negative control antibody in RAST (vi). (B) Synovial tissue, primary rheumatoid arthritis synovial fibroblast cells (RASFC) and human dermal endothelial cells (HDEC) were assessed for F-actin and for focal adhesions by staining for vinculin and p-paxillin by fluorescent microscopy. F-actin and 4′,6-diamidino-2-phenylindole nuclear staining in synovial tissue from RA (i), osteoarthritis and control synovium. Weak F-actin staining was observed in healthy control tissue (ii). Representative images of F-actin staining in RASFC (iii) and HDEC (iv). Positive staining of cells for focal adhesion complexes as evidenced by vinculin staining localised primarily at the periphery of RASFC cells (v). Strong p-paxillin staining in HDEC is shown in (vi) with a punctate staining pattern, where localisation of p-paxillin was observed throughout the cells in addition to the polarised, leading edge of the cell.
F-actin staining was observed in RA, osteoarthritis and control synovial tissue. F-actin was expressed throughout RAST with dramatically stronger staining intensity in blood vessels (figure 1B(i)) compared with normal control tissue (figure 1B(ii)). Figure 1B(iii), (iv) shows representative images of F-actin staining in RASFC and HDEC demonstrating intact bundles of stress fibres orientated along the cell in a unidirectional manner. Vinculin staining localised primarily at the periphery of RASFC (v) and strong punctate p-paxillin staining was demonstrated throughout the cell in HDEC (vi), and at the polarised leading cell edge. High powered images of F-actin on synovial tissue blood vessels in RA, osteoarthritis and normal controls are demonstrated in supplementary figure 1, available online only. Strong F-actin staining was demonstrated in RA (A–D) and in osteoarthritis (E, F) synovial tissue, but weak staining was shown in normal control tissue (G, H). F-actin fibres and abnormal blood vessel morphology were visible in RAST (A–D).
A-SAA-stimulated integrin binding and cytoskeletal rearrangement
A-SAA significantly increased cell surface β1 binding to anti-integrin-coated plates in RASFC, resulting in a 2.5-fold increase compared with unstimulated cells (p=0.02) (figure 2A(i)). A-SAA induced a fourfold increase in RASFC surface αvβ3 binding to coated plates compared with control cells (p<0.05) (figure 2A(ii)).

Serum amyloid A (A-SAA) regulates integrin expression and cytoskeletal rearrangement. Control and A-SAA (10 μg/ml) treated rheumatoid arthritis synovial fibroblast cell (RASFC) were examined for integrin binding by specific adhesion assay (n=5). A-SAA significantly increases (A) β1 (i) integrin expression and αvβ3 (ii) integrin expression. (B) A-SAA induces cytoskeletal rearrangement in RASFC (upper panels) and human dermal endothelial cell (lower panels) compared with basal control as evidenced by the formation of membrane ruffling (arrow) and loss of focal adhesions (vinculin) (red staining, lower panels). Original magnification ×40.
Figure 2B demonstrates the effect of A-SAA on cytoskeletal architecture and cell shape. A-SAA induced actin cytoskeleton disassembly and loss of focal adhesions in both RASFC and HDEC evidenced by the dissolution of intact stress fibres and loss of punctate vinculin staining in A-SAA-treated cells compared with controls. Separate images are also demonstrated in supplementary figure 2, available online only. In addition, A-SAA stimulation induced filopodial and lamellipodial-like structures, thin actin-rich extensions of the cytoplasm and membrane ruffling, key cellular changes during directed cell migration.
At a functional level A-SAA-induced HDEC invasion and wound repair were inhibited in the presence of anti-β1 (figure 3) with no effect observed for the IgG negative control.

Serum amyloid A (SAA) induced invasion and migration is mediated partly by β1 integrin. (A) SAA-induced invasion in human dermal endothelial cell (HDEC) was decreased upon co-incubation with anti-β1 integrin. (B) Quantification of HDEC invasion. (C) Rheumatoid arthritis synovial fibroblast cell migration is increased by SAA, as evidenced by cells repopulating the wound, but partly inhibited upon SAA/anti-β1 integrin co-stimulation.
A-SAA upregulates GTPase expression in vitro
A-SAA (50 μg/ml) significantly increased the detection of active Cdc42, without a change in global expression at 6 h in both SFC (figure 4A) and HDEC (figure 4B) (p<0.05); however, no significant increase in A-SAA-induced detection of active Cdc42 expression was observed at 12 h. Conversely, no significant increase in the active GTP-bound RhoA, or global expression was observed following A-SAA stimulation (figure 4C,D). Although not significant, RhoA appeared downregulated at 12 h.

Serum amyloid A (A-SAA) induces ρ-GTPase activity in K4 synoviocytes and human dermal endothelial cell (HDEC). Representative western blots for Cdc42 and RhoA following 6 and 12 h A-SAA (10 μg/ml) and tumour necrosis factor alpha (TNFα; 10 ng/ml) stimulation. A-SAA upregulates Cdc42 expression in (A) K4 synoviocytes and (B) HDEC, which was significantly greater compared with controls. A-SAA induction of ρ expression in (C) K4 synoviocytes and (D) HDEC. Quantification of the active forms of Cdc42 and ρ proteins are displayed as bar charts underneath representative blots.
A-SAA-induced cell growth, migration and invasion
To examine if A-SAA-induced cell growth, angiogenesis, migration/invasion are mediated through integrin-ρ-GTPase signalling pathways, cells were stimulated with A-SAA in the presence or absence of small molecular weight inhibitors to Rac1 (NSC23766) and Cdc42 (187-1).
A-SAA induced a threefold increase in RASFC growth (p<0.05), an effect significantly blocked in the presence of NSC23766 and 187-1 (figure 5A). A-SAA induced HDEC tube formation after 24 h from a basal level of 33.2±7.67 to 44.2±7.78, an effect reduced upon Rac1 and Cdc42 inhibition (30.66±9.24 and 30.7±6.52, respectively) (figure 5B). A-SAA-induced HDEC invasion was inhibited in the presence of NSC23766 and 187-1 (figure 5C). NSC23766, 187-1 alone or DMSO vehicle control had minimal effects on cell growth, angiogenesis, invasion or cell viability.

Serum amyloid A (A-SAA)-induced proliferation, invasion and tubule formation are Rac and Cdc42 dependent. (A) A-SAA-induced cell growth is decreased in the presence of Rac and Cdc42 inhibitors (NSC23766 and 187-1). (B) Representative photomicrograph showing human dermal endothelial cell (HDEC) invasion following A-SAA (10 μg/ml) stimulation. A-SAA-induced invasion of HDEC is markedly decreased upon co-incubation with 187-1 and NSC23766. (C) Representative photomicrographs of HDEC tubule formation on Matrigel matrix following 24 h of incubation with A-SAA (10 μg/ml) alone and in the presence of 187-1 and NSC23766.
A-SAA induced wound closure in RASFC, an effect that was markedly decreased with NSC23766 (figure 6A) and 187-1 (figure 6C) in which RASFC failed to repopulate the wound. Furthermore, NSC23766 and 187-1 inhibited A-SAA-induced actin disassembly and filopodial formation in RASFC was similar to basal suggesting that A-SAA-induced cell movement is mediated by Cdc42 and Rac1 (figure 6B,D). No effect was observed with inhibitors alone or DMSO vehicle control.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Serum amyloid A (A-SAA)-induced cell migration is Rac and Cdc42 dependent. (A) Representative photomicrograph showing rheumatoid arthritis synovial fibroblast cell (RASFC) repopulating the wound in response to A-SAA (10 μg/ml), but neutralisation of Rac prevented wound closure even in the presence of A-SAA. (B) The effect of Rac inhibition (NSC23766) on the integrity of the actin cytoskeleton was investigated by immunofluorescence following 24 h stimulation. Intact stress fibres are shown by white arrows in basal cells. A-SAA alone induced actin disassembly and the formation of lamellipodia (red arrows) compared with basal control, while Rac inhibition enabled actin formation. Furthermore, Rac blockade prevented A-SAA-induced actin disassembly (×20 magnification). (C) A-SAA alone induces RASFC migration but neutralisation of Cdc42 prevented A-SAA-induced wound closure. (D) A-SAA treated cells underwent actin cytoskeleton disassembly. Co-incubation with a Cdc42 inhibitor (187-1) failed to initiate actin re-distribution. No effect was observed with inhibitor alone or dimethyl sulfoxide vehicle control.
Discussion
RA is a chronic inflammatory disease characterised by increased cell migration and synovial hyperplasia. Migration is a multistep process regulated by integrin–FAK-ρ-GTPase-mediated pathways.26 27 We demonstrated localised αvβ3 and β1-integrin expression and actin cytoskeletal proteins in RAST and cells. We showed that A-SAA significantly induces binding of αvβ3 and β1 and actin disassembly in RASFC and HDEC. A-SAA-induced cytoskeletal rearrangement results in the differential regulation of Cdc42 and RhoA activity. Finally, we demonstrated that A-SAA-induced cell growth, angiogenesis, invasion and migration are partly mediated through cytoskeletal dynamics and ρ-GTPase activation.
In this study, αvβ3 and β1 integrin expression was predominantly localised to the perivascular regions, with some lining layer expression, consistent with previous studies showing expression on RAST blood vessels, particularly at an early stage of neoangiogenesis.28 Integrin expression in RA synovial membrane is closely associated with the expression pattern of A-SAA and its receptors, FRPL-1, scavenger receptor class B1 and TLR2.5 7 8 Several in-vitro studies have shown a key role for αvβ3 in angiogenesis, osteoclast-mediated bone resorption, matrix metalloproteinase production and X linked inhibitor of apoptosis protein (XIAP) expression.29,–,32 In-vivo animal studies have also demonstrated significant reductions in synovial angiogenesis, cell infiltration and subsequent pannus formation following αvβ3 blockade.29 β1-integrin is involved in angiogenesis and in the response of T cells to fibronectin-induced chemotactic signals33 and the attachment of RASFC to ECM components and subsequent invasion of cartilage.21 25 In the current study, we demonstrated F-actin staining in the lining layer and perivascular regions, with strong expression of F-actin, p-paxillin and vinculin in RASFC and HDEC. To date few studies have examined the expression of these proteins in RA cells; however, Shahrara et al34 showed significantly higher expression of p-FAK in RAST compared with osteoarthritis or controls. Furthermore, p-FAK was expressed on synovial tissue endothelial cells and lymphocytes, whereas p-paxillin was expressed on RAST lining.34
Our previous studies demonstrated increased A-SAA in the RA joint and showed that it is critically involved in synovial tissue cell migration and angiogenesis in vitro, ex vivo and in vivo. To probe further whether A-SAA plays a critical role in driving these processes we examined its effect on integrin–Fak mediated pathways. We showed that A-SAA significantly upregulates cell surface αvβ3 and β1-integrin binding in RASFC and has profound effects on the integrity of the actin cytoskeleton, by F-actin dissolution and complete loss of focal adhesions. We demonstrated that A-SAA induced microspike configuration and cell protrusion via formation of filopodia, probably increasing cell motility in RASFC and HDEC. In this study we demonstrated that A-SAA-induced angiogenesis, cell migration and invasion are inhibited by anti-β1 integrin. Whereas studies have demonstrated interactions of FPRL-1 and TLR2 receptors with integrin/Fak pathways,35,–,37 this is the first evidence of a direct effect of A-SAA on integrin signalling pathways. Previous studies have shown interactions between specific forms of A-SAA and ECM glycosaminoglycans and proteoglycans, facilitating the binding of A-SAA to basement membrane.38 ECM can bind integrins in addition to other ECM components such as heparin sulphate and collagen,39 40 suggesting that A-SAA interactions with the ECM may mediate a diverse array of biological processes. Furthermore, studies have shown that mast cell adhesion to an immobilised A-SAA–ECM complex occurs through integrin recognition, possibly via β1-integrin activation.41 Moreover, Preciado-Patt et al42 demonstrated A-SAA interactions with ECM, laminin or vitronectin-induced adhesion of resting CD4 cells in a β1-integrin-dependent manner.
ρ-GTPases regulate the assembly and organisation of the actin cytoskeleton through coordinated but distinct processes.43 Rac-1 and Cdc42 promote membrane protrusion and induce the formation of lamellipodia and filopodia, respectively, whereas RhoA inhibits such protrusions, instead promoting myosin contractility, resulting in the formation of stress fibres and focal adhesions.44 In this study, we demonstrated that A-SAA significantly increased Cdc42 activity, with no effect on RhoA, suggesting A-SAA induces membrane protrusion with a concomitant downregulation of stress fibre formation. Functionally, we demonstrated that A-SAA-induced RASFC growth and cell migration were inhibited by specific Rac-1 and Cdc42 inhibitors. In addition, NSC23766 and 187-1 inhibited A-SAA-induced endothelial cell tube formation and invasion, suggesting a role in blood vessel formation. Cdc42 and Rac-1 are known to regulate endothelial cell morphology and are required for the process of endothelial cell intracellular vacuole formation and coalescence, whereas RhoA is involved in endothelial cell branching/sprouting events.17 45 In the context of inflammatory arthritis, several studies have shown differential blood vessel shape and patterns in the joint with RA characterised by straight vessels with regular branching, while Psoriatic Arthritis has elongated, tortuous vessels with no branching.46 47
A-SAA failed to induce reorganisation of the actin cytoskeleton in the presence of Rac-1 and Cdc42 inhibition, thus A-SAA effects cell behaviour and shape through integrin-ρ-GTPase-linked signalling mechanisms. This is consistent with previous studies using RASFC in which the inhibition of small GTPases and Fak signalling components inhibits cell migration and invasion.48 In inflammatory bowel disease, ρ-GTPases modulate proinflammatory cytokine production via nuclear factor kappa B activation,49 and similarly RhoA-mediated signalling promotes nuclear factor kappa B activation, proinflammatory cytokine production and proliferation in RASFC.50,–,52 In vitro an inhibitor of F-actin polymerisation, latrunculin-A, decreased migration and proliferation of SFC from human TNF transgenic mice compared with wild-type SFC.52 Finally, several studies have shown that lipoxin A4, which competes with A-SAA for the FPRL-1 receptor, can inhibit these processes,37 53 further supporting a role for A-SAA-induced cytoskeletal pathways. In conclusion, this is the first study to show that A-SAA-induced cell migration and invasion is mediated through ρ-GTPases and altered cytoskeletal dynamics.
References
Supplementary materials
Web Only Data
Files in this Data Supplement:
Footnotes
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval This study was conducted in accordance with the Declaration of Helsinki, and was approved by the St Vincent's University Hospital ethics committee.
-
Provenance and peer review Not commissioned; externally peer reviewed.