Article Text

Abstract
Objectives Genetic variants in the transcription factor STAT4 are associated with increased susceptibility to systemic lupus erythematosus (SLE) and a more severe disease phenotype. This study aimed to clarify how the SLE-associated intronic STAT4 risk allele rs7574865[T] affects the function of immune cells in SLE.
Methods Peripheral blood mononuclear cells (PBMCs) were isolated from 52 genotyped patients with SLE. Phosphorylation of STAT4 (pSTAT4) and STAT1 (pSTAT1) in response to interferon (IFN)-α, IFN-γ or interleukin (IL)-12, total levels of STAT4, STAT1 and T-bet, and frequency of IFN-γ+ cells on IL-12 stimulation were determined by flow cytometry in subsets of immune cells before and after preactivation of cells with phytohaemagglutinin (PHA) and IL-2. Cellular responses and phenotypes were correlated to STAT4 risk allele carriership. Janus kinase inhibitors (JAKi) selective for TYK2 (TYK2i) or JAK2 (JAK2i) were evaluated for inhibition of IL-12 or IFN-γ-induced activation of SLE PBMCs.
Results In resting PBMCs, the STAT4 risk allele was neither associated with total levels of STAT4 or STAT1, nor cytokine-induced pSTAT4 or pSTAT1. Following PHA/IL-2 activation, CD8+ T cells from STAT4 risk allele carriers displayed increased levels of STAT4 resulting in increased pSTAT4 in response to IL-12 and IFN-α, and an augmented IL-12-induced IFN-γ production in CD8+ and CD4+ T cells. The TYK2i and the JAK2i efficiently blocked IL-12 and IFN-γ-induced activation of PBMCs from STAT4 risk patients, respectively.
Conclusions T cells from patients with SLE carrying the STAT4 risk allele rs7574865[T] display an augmented response to IL-12 and IFN-α. This subset of patients may benefit from JAKi treatment.
- STAT4
- IL-12
- interferon
- systemic lupus erythematosus
- JAK inhibitors
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Systemic lupus erythematosus (SLE) is an autoimmune rheumatic disease with a strong genetic component.1 Genetic association studies have identified >80 loci associated with an increased susceptibility to SLE.2 One of the strongest SLE risk loci outside the HLA region is signal transducer and activator of transcription (STAT)4, with the most significantly associated single-nucleotide polymorphisms (SNPs) in STAT4 being located in the third intron of the gene.3–5 However, there is a large linkage disequilibrium (LD) block extending towards the 3′ end of the gene and the causal SNP has not been inferred.6 Since STAT1 is located adjacent to STAT4, it is possible that the risk SNPs found in STAT4 have a regulatory effect on STAT1.
Both STAT4 and STAT1 encode transcription factors of importance to the immune system. STAT4, mainly expressed in T cells and NK cells, is required for the inflammatory response following IL-12 receptor (IL-12R) stimulation.7–9 STAT4 is also involved in the non-canonical signalling pathway of the type I interferon (IFN) receptor (IFNAR).10 11 On IL-12R engagement, STAT4 is phosphorylated by JAK2 and TYK2, leading to dimerisation and translocation to the nucleus, where it binds DNA and induces the expression of a large number of proinflammatory genes, including IFNG.12 STAT1 is widely expressed and part of the canonical signalling pathways for the type I IFN receptor (IFNAR) and the IFN-γ receptor (IFNGR).13 Increased expression and activation of STAT1 has been reported in patients with lupus, which support the important role of the IFN system in SLE.14
The SLE-associated STAT4 SNPs are linked to a more severe disease phenotype with an earlier onset of disease, and an increased risk for stroke and nephritis with severe renal insufficiency.4 15–17 Despite the strong association of STAT4 risk SNPs with rheumatic diseases and clinical subphenotypes, very little is known about the molecular mechanisms whereby these risk gene variants contribute to autoimmune disease. Previous studies have reported increased expression of STAT4 mRNA in osteoblasts4 and peripheral blood mononuclear cells (PBMCs) from healthy individuals18 and patients with SLE19 carrying the STAT4 risk allele. Furthermore, patients with SLE carrying the STAT4 risk allele have an increased expression of IFN-induced genes in peripheral blood cells despite having a decreased IFN-α activity in sera.20 In the present study, we set out to define how the STAT4 risk variant affects cellular functions in different types of immune cells in SLE. Using primary cells from genotyped patients with SLE, we examined STAT4-dependent and STAT1-dependent cellular functions on the single-cell level in unstimulated and in vitro stimulated cells and correlated these to rs7574865, which is one of the strongest SLE-associated SNPs in STAT4. Finally, Janus kinase (JAK) inhibitors were evaluated in vitro for their capacity to restore augmented cellular responses in PBMCs from patients with SLE carrying the STAT4 risk allele.
Patients and methods
Patients and cells
Peripheral blood was sampled from 52 patients with SLE, fulfilling ≥4 of the 1982 American College of Rheumatology (ACR) classification criteria,21 at the Rheumatology clinic, Uppsala University Hospital. Disease activity and organ damage were measured with the SLE Disease Activity Index 2000 (SLEDAI-2K)22 and the Systemic Lupus International Collaborating Clinics/ACR Damage Index,23 respectively. Patients were genotyped with the Immunochip or the Infinium OmniExpressExome-8 V.1.4 chip (Illumina) and stratified by rs7574865 (NC_000002.12:g.191099907T>G). PBMCs were prepared by Ficoll density-gradient centrifugation and viability frozen in liquid nitrogen. In some experiments, PBMCs were preactivated with 1.5% phytohaemagglutinin (PHA; Life Technologies) and 2.5 ng/mL IL-2 (Miltenyi) for 72 hours. After washing with phosphate buffered saline, cells were rested 4 hours before re-stimulation. IFN-α and IFN-γ levels in plasma were determined with an in-house immunoassay24 and the human IFN-γ ELISA MAX Standard kit (Biolegend), respectively.
Phosphorylation of STAT4 and STAT1
PBMCs were stimulated with IFN-α2b (IntronA; Schering-Plough), IFN-γ or IL-12 (both Peprotech) at 37°C. Cells were fixed with 2% PFA, permeabilised with Perm Buffer III (BD Biosciences) and stained for cell surface markers (online supplementary table 1). STAT4 and STAT1 protein levels were determined using rabbit polyclonal anti-STAT4 IgG (C20; Santa Cruz) together with PE-labelled F(ab′)2 donkey anti-rabbit IgG or an anti-STAT1 mAb (1/Stat1; BD Biosciences). The geometric mean fluorescence intensity (G-MFI) of STAT4 and STAT1 was divided by the G-MFI of STAT4/STAT1-negative cells. Phospho-specific antibodies to STAT4 (pY693, 38p-Stat4) and STAT1 (pY701, 4a, both BD Biosciences) were used to assess phosphorylation. Cytokine-induced phosphorylation was determined by subtracting the G-MFI of non-stimulated cells from the G-MFI of stimulated cells. Maximal phosphorylation of STAT4 (pSTAT4) and STAT1 (pSTAT1) was detected after 20 min (online supplementary figure S1A,B). EC50 values were determined in resting and PHA/IL-2 preactivated cells (IFN-α 200 U/mL, IFN-γ 0.1 ng/mL and IL-12 1 ng/mL; PHA/IL-2 activated cells IFN-α 500 U/mL and IL-12 5 ng/mL; online supplementary figure S1C,D). Unless otherwise stated, a dose of cytokine corresponding to 10 times the EC50 dose was used.
Supplementary file 1
IFN-γ production
PHA/IL-2 preactivated cells were re-stimulated with 5 ng/mL IL-12 for 15 hours or 20 ng/mL phorbol 12-myristate-13-acetate (PMA) and 1 ng/mL of the calcium-ionophore A23187 (both Sigma Aldrich) for 6 hours, in the presence of GolgiPlug (BD Biosciences) the last 12 and 5 hours, respectively. Cells were permeabilised with saponin and stained with an anti-IFN-γ (B27) or an isotype control (MOPC-21, both BD Biosciences) mAb. IL-12-specific production of IFN-γ was determined by subtracting the frequency of IFN-γ+ cells in unstimulated cells.
T-bet expression
T-bet expression was determined using anti-T-bet (O4-46) mAb and the Foxp3 Transcription Factor Staining Buffer (eBiosciences).
Janus kinase inhibitors
The pan-JAK inhibitor (pan-JAKi) tofacitinib,25 26 the JAK2 selective inhibitor BMS-911543 (JAK2i)27 and the TYK2 selective inhibitor compound 35 (TYK2i)28 (all provided by AstraZeneca) were added to cell cultures at indicated concentrations 20 min prior to cytokine stimulation.
Flow cytometry
Flow cytometry data were collected on a FACSCantoII instrument with FACSDiva software V.7.0 (BD Biosciences). Data were analysed using FlowJo software V.10.0.8 (Tree Star).
Statistics
Inter-experiment normalisation was performed as detailed in online supplementary methods. Comparisons of clinical characteristics were performed with the χ2 or Kruskal-Wallis test. For comparisons of three genotypes, a linear regression model assuming an additive effect was used. No age effect was observed and consequently age was not included as a covariate. For comparisons of two groups, the Mann-Whitney U test was used. Statistical tests were two-tailed. IC50 values for JAK inhibitors were determined with a four-parameter logistic regression model. Statistical calculations were performed with GraphPad Prism V.6.02 or IBM SPSS Statistics V.25.
Results
PHA/IL-2-activated T cells from patients with SLE carrying the STAT4 risk allele have an increased pSTAT4 in response to IL-12 and IFN-α
To study whether rs7574865[T] (hereafter referred to as the STAT4 risk allele) is associated with alterations in STAT4-dependent or STAT1-dependent cellular traits, we isolated PBMCs from 52 genotyped patients with SLE. All patients had low disease activity (SLEDAI-2K≤4), and the three groups were clinically well matched (table 1).
Patient characteristics
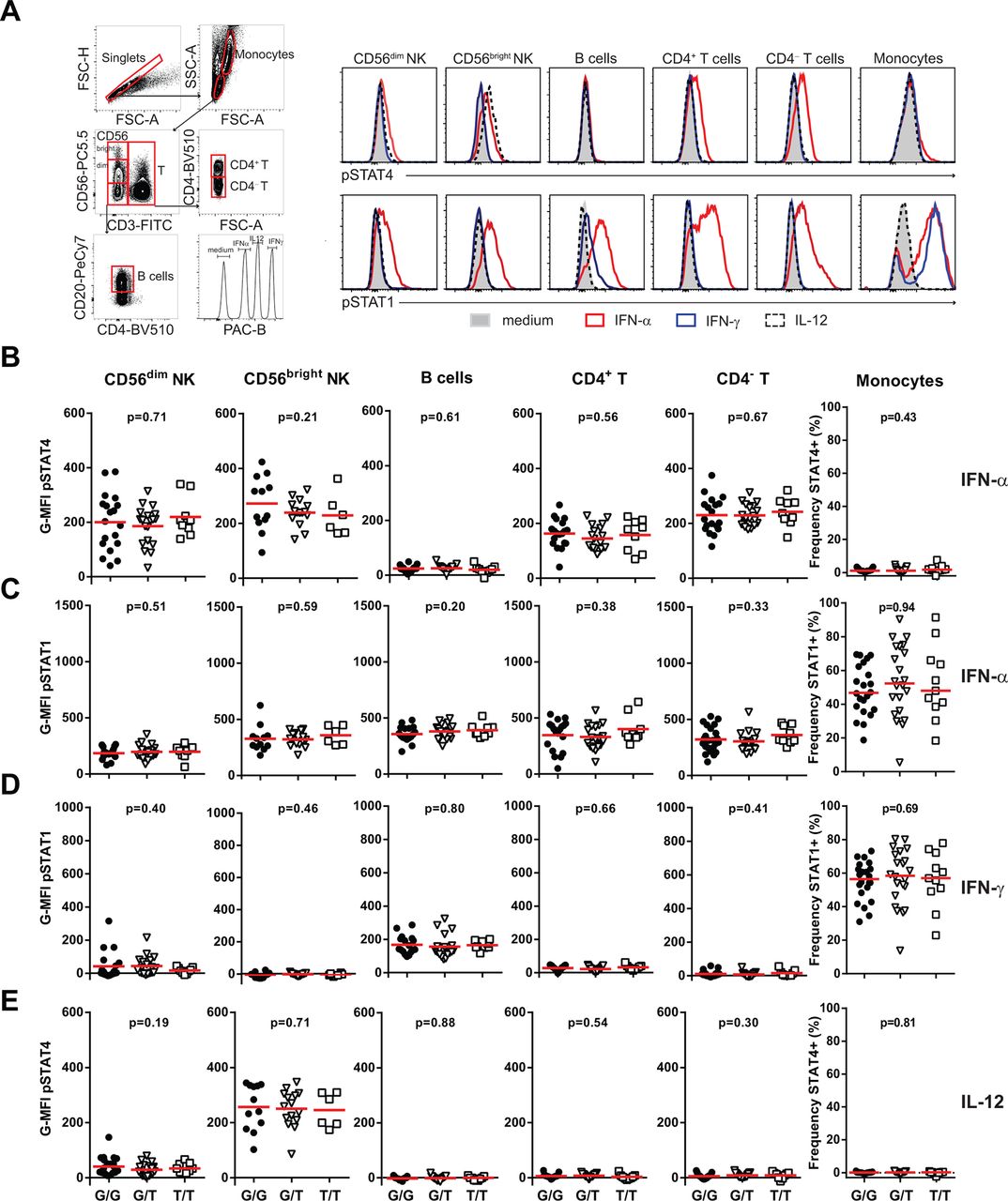
Initially, the phosphorylation of STAT4 (pSTAT4) and STAT1 (pSTAT1) was determined in CD56dim NK cells, CD56bright NK cells, B cells, CD4+ T cells, CD4– T cells and monocytes, before and after IFN-α, IFN-γ or IL-12 stimulation. The STAT4 risk allele was not correlated to basal levels (data not shown), nor IFN-α, IFN-γ-induced or IL-12-induced pSTAT4 or pSTAT1 in any cell type studied (figure 1A–E). Furthermore, total protein levels of STAT4 and STAT1, and the IFNAR and IFNGR expression were not correlated to the STAT4 risk allele (online supplementary figure S2).

The STAT4 risk allele does not affect IFN-α, IFN-γ or IL-12-induced phosphorylation of STAT4 or STAT1 in resting peripheral blood mononuclear cells (PBMCs). PBMCs from patients with systemic lupus erythematosus (SLE) were stimulated with 2000 U/mL IFN-α (A–C), 1 ng/mL IFN-γ (A, D) or 10 ng/mL IL-12 (A, E) for 20 min and phosphorylation of STAT4 (pSTAT4) and STAT1 (pSTAT1) was determined in indicated cell population, by flow cytometry. (A) Flow cytometry plots with gating strategy from one representative donor. (B–E) Cumulative data from 19 homozygous protective (G/G, black circles), 21 heterozygous (G/T, open triangles) and 9 homozygous risk (T/T, open squares) STAT4 patients with SLE. For monocytes, frequencies of pSTAT4+ and pSTAT1+ cells are shown because of the bimodal distributions. Horizontal red bars indicate the mean value. Due to <100 acquired events, data from CD56bright NK cells and B cells are from 12 G/G, 16 G/T and 6 T/T and 17 G/G, 21 G/T and 9 T/T individuals, respectively.
To further study IL-12 responsiveness in T cells, IL-12R expression was induced by pre-stimulation of cells with PHA and IL-2 for 72 hours.29 Following re-stimulation with IL-12, an increased pSTAT4 was evident in CD8+ T cells from STAT4 risk allele carriers (P=0.003, figure 2A,B). This effect was confined to the CD45RA+CD57– naïve and the CD45RA–CD57– memory CD8+ T cells (P=0.02 and P=0.006, respectively, figure 2C) and the effect was risk allele dosage–dependent with heterozygous STAT4 risk allele patients displaying intermediate levels of pSTAT4. A slightly increased pSTAT4 was also seen in response to IL-12 in the CD45RA–CD57– memory CD4+ T cells (P=0.07, figure 2C). Importantly, current medication (figure 2D), plasma levels of IFN-α (P=0.66) or disease activity (P=0.37) were not associated with the IL-12-response, demonstrating that the increased pSTAT4 in these STAT4 risk allele carriers was not secondary to disease or therapy. Furthermore, analysis of cells sampled almost 2 years apart showed that the IL-12-induced pSTAT4 in CD8+ T cells was a relatively stable phenotype (figure 2E).

PHA/IL-2 preactivated T cells from patients with systemic lupus erythematosus (SLE) carrying the STAT4 risk allele have an increased phosphorylation of STAT4 in response to IL-12. PHA/IL-2 pre-activated peripheral blood mononuclear cells were stimulated with 50 ng/mL IL-12 for 20 min. Phosphorylation of STAT4 (pSTAT4) was determined in indicated cell populations using flow cytometry. (A) Histograms from one representative donor. (B–D) Cumulative data from 19 homozygous protective (G/G, black circles), 20 heterozygous (G/T, open triangles) and 9 homozygous risk (T/T, open squares) STAT4 patients with SLE. Due to <100 acquired events, data from CD8+ effector and CD4+ naïve T cells in (C) are from 17 G/G, 19 G/T and 8 T/T patients with SLE. (D) pSTAT4 in CD8+ T cells from patients stratified for current medication with hydroxychloroquine (HCQ), prednisolone (Pred), azathioprine (AZA), mycophenolate mofetil (MMF) or methotrexate (MTX). (E) pSTAT4 in CD8+ T cells from seven patients sampled twice at indicated time intervals. Horizontal red bars indicate the mean value. *P<0.05 and **P<0.01.
In PHA/IL-2-preactivated PBMCs, slightly increased pSTAT4 and pSTAT1 was also seen in IFN-α-stimulated CD8+ T cells (P=0.04 and P=0.10) from STAT4 risk allele carriers, but not in CD4+ T cells (P=0.36 and P=0.22) or NK cells (P=0.19 and 0.34, online supplementary figure S3).
In conclusion, PHA/IL-2 preactivated CD8+ T cells from patients with SLE carrying the STAT4 risk allele have an increased response to IL-12 and IFN-α. Since the strongest effect was seen in IL-12-stimulated cells, we focused our further studies on the IL-12 response.
T cells from patients with SLE carrying the STAT4 risk allele have increased levels of STAT4 after PHA/IL-2 activation
The increased IL-12-induced pSTAT4 may reflect increased levels of STAT4 or increased IL-12R expression. Analysis of total levels of STAT4 in PHA/IL-2 preactivated CD45RA–CD57– memory CD8+ T cells showed a strong correlation with IL-12-induced pSTAT4 (P=0.002, figure 3A). In contrast to resting T cells, PHA/IL-2 preactivated cells from STAT4 risk allele carriers had increased levels of STAT4 in the CD45RA+CD57– naïve (P=0.003) and the CD45RA–CD57– memory (P=0.01) CD8+ T cells and slightly increased levels in CD45RA+CD57– naïve (P=0.06) and CD45RA–CD57– memory (P=0.09) CD4+ T cells (figure 3B).

T cells from STAT4 risk patients with systemic lupus erythematosus (SLE) have increased levels of STAT4 following PHA/IL-2 stimulation. Total protein levels of STAT4 and expression of the IL-12 receptor specific subunit IL12RB2 was determined in PHA/IL-2 preactivated cells in indicated cell populations by flow cytometry. (A, C) Correlation of total levels of STAT4 (A) or IL12RB2 (C) expression with IL-12-induced phosphorylation of STAT4 (pSTAT4) in CD8+ memory T cells. (B, D) Total levels of STAT4 (B) and expression of IL12RB2 (D) in indicated cell populations stratified by STAT4 genotype. Data from 14 homozygous protective (GG, black circles), 9 heterozygous (G/T, open triangles) and 8 homozygous risk (TT, open squares) STAT4 patients with SLE. Due to <100 acquired events, data from CD8+ effector T cells are from 12 G/G, 8 G/T and 7 T/T patients with SLE. Horizontal red bars indicate the mean value. *P<0.05 and **P<0.01.
In terms of IL-12R expression, there were no associations between IL12RB2 expression and IL-12-induced pSTAT4 (figure 3C) or between levels of IL12RB2 and STAT4 genotype (figure 3D).
Thus, patients with STAT4 risk allele have an augmented induction of STAT4 protein following PHA/IL-2 stimulation, which may possibly explain the increased response to IL-12.
T cells from patients with SLE carrying the STAT4 risk allele have an increased production of IFN-γ in response to IL-12
As IL-12-induced IFN-γ production is mediated via STAT4 activation, we next analysed whether the STAT4 risk allele was associated with increased IFN-γ production. pSTAT4 levels in IL-12-stimulated CD8+ and CD4+ T cells were associated with the frequency of IFN-γ+ cells (P=0.007 and P=0.04, respectively), and consistent with the phosphorylation data, CD45RO+CD57– memory CD8+ T cells from risk allele carriers had a higher frequency of IFN-γ+ cells (P=0.03, figure 4A). Increased IFN-γ production was also observed in the CD45RO+CD57– memory CD4+ T cells (P=0.06). A general increased IFN-γ production in T cells from STAT4 risk individuals was excluded by the fact that PMA-induced IFN-γ production was not increased (figure 4B). Despite a risk-allele dosage-dependent numerical increase in IFN-γ plasma levels, this difference was not statistically significant (P=0.32, figure 4C).

PHA/IL-2 preactivated T cells from STAT4 risk patients with systemic lupus erythematosus (SLE) have an increased IFN-γ production in response to IL-12. (A, B) PHA/IL-2 activated peripheral blood mononuclear cells were re-stimulated with 5 ng/mL IL-12 for 15 hours (A) or 20 ng/mL PMA together with 1 ng/mL A23187 for 6 hours (B). The frequency of IFN-γ+ cells was determined in CD8+ and CD4+ T cells and subsets thereof using flow cytometry. Data from 18 STAT4 homozygous protective (G/G, black circles), 21 heterozygous (G/T, open triangles) and 9 homozygous risk (T/T, open squares) patients with SLE. Data of CD8+ T cells from one G/T individual (indicated as a filled triangle in A) was considered an outlier and was excluded from the statistical analysis of CD8+ T cells and subsets thereof. Due to <100 acquired events, data from one G/G and two G/T from the CD8+ naïve T cells, one G/T and two T/T from the CD4+ naïve T cells, and one G/G from the CD4+ memory T cells were excluded. (C) The concentration of IFN-γ was determined in plasma from 19 G/G, 19 G/T and 10 T/T patients with SLE. Measurements below the detection limit (7 pg/mL, n=7) were assigned a value of half the detection limit. Values above the detection limit (1000 pg/mL, n=4) were assigned a value of 1000 pg/mL. (D) The frequencies of T-bet+ CD4+ T cells in PHA/IL-2 preactivated cells were determined in 12 G/G, 11 G/T and 9 T/T patients with SLE. (A–D) Horizontal red bars indicate the mean value. *P<0.05.
The STAT4 risk allele did not appear to affect Th1 differentiation as neither the frequency of IFN-γ+ (figure 4B) nor T-bet+ (figure 4D) CD4+ T cells correlated with STAT4 genotype.
Thus, T cells from patients with SLE carrying the STAT4 risk allele have an augmented IL-12-induced production of IFN-γ.
JAK inhibitors block the IL-12 response in STAT4 risk patients with SLE
To investigate the possibility of pharmacological modulation of the IL-12/IFN-γ axis in STAT4 risk patients, we compared the effect of the pan-JAKi tofacitinib, with two JAKis selectively targeting JAK2 or TYK2. Dose titrations of the JAKis in IL-12, IFN-α or IFN-γ-stimulated cells from healthy individuals revealed that the TYK2i was most selective for IL-12 blockade (figure 5A,B). As expected, the TYK2i also blocked IFN-α-induced activation of cells, whereas the JAK2i blocked IFN-γ-induced activation and the pan-JAKi blocked both IFN-γ-induced and IFN-α-induced activation of cells. Importantly, cells from SLE patients carrying the STAT4 risk allele were equally sensitive to TYK2i-mediated inhibition of IL-12-induced pSTAT4 and IFN-γ production as cells from non-risk carriers and healthy donors (figure 5C,D). Likewise, the JAK2i and pan-JAKi efficiently blocked IFN-γ signalling in patients with SLE carrying the STAT4 risk allele (figure 5E). Collectively, these data demonstrate the feasibility to target the IL-12/IFN-γ axis with JAKis in STAT4 risk allele carriers.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Janus kinase inhibitors block the IL-12 and IFN-γ response in peripheral blood mononuclear cells (PBMCs) from STAT4 risk patients with systemic lupus erythematosus (SLE). (A) Healthy donor PBMCs were treated with serial dilutions of the JAK2 selective (JAK2i), the TYK2 selective (TYK2i) and the pan-JAK inhibitor (pan-JAKi) before being stimulated with 5 ng/mL IL-12, 0.1 ng/mL IFN-γ or 200 U/mL IFN-α for 20 min. IL-12-stimulated cells were preactivated with PHA/IL-2. Phosphorylation of STAT4 (pSTAT4) and STAT1 (pSTAT1) was determined by flow cytometry in IL-12-stimulated cells and IFN-γ or IFN-α-stimulated cells, respectively. Data (mean±SD of two individuals) from IL-12 and IFN-α-stimulated cells are shown for CD3+ T cells and from IFN-γ-stimulated cells for monocytes. (B) IC50 values for each inhibitor. (C–E) PBMCs from healthy donors (HD), patients with SLE homozygous for the protective STAT4 allele (G/G) and patients with SLE carrying one or two STAT4 risk alleles (G/T+T/T) were incubated with 200 nM TYK2i (C, D), 70 nm JAK2i or 30 nM pan-JAKi (E). (C, D) Inhibition of IL-12-induced pSTAT4 in CD8+ T cells (C) and reduction in the frequency of IFN-γ+ memory CD45RO+CD57– CD8+ T cells (D). (E) Inhibition of IFN-γ-induced pSTAT1 in monocytes. (C–E) Open triangles and squares denote G/T and T/T patients with SLE, respectively.
Discussion
Here, we report the first evidence of how an intronic SNP in the major SLE risk locus STAT4 affects the function of immune cells from patients with SLE in a cell-type-specific and context-dependent manner. We specifically selected patients in remission, or with low disease activity, to avoid effects on the cells due to a prominent ongoing in vivo immune activation. Initially, we analysed pSTAT4 and pSTAT1 in primary immune cells from patients with SLE following IFN-α, IFN-γ and IL-12 stimulation, three cytokines elevated in sera from patients with SLE.30–32 In resting PBMCs, there was no association of STAT4 genotype with IFN-α, IFN-γ or IL-12-induced pSTAT4 or pSTAT1. However, re-stimulation of PHA/IL-2 preactivated cells with IL-12 or IFN-α induced a risk-allele dosage-dependent increased pSTAT4 in CD8+ T cells.
The increased IL-12-induced pSTAT4 in CD8+ T cells from STAT4 risk individuals was accompanied by an augmented IFN-γ production, mainly confined to the CD45RO+CD57– memory subset. Increased IFN-γ production was also seen in IL-12-stimulated CD4+ T cells from STAT4 risk individuals. The augmented IFN-γ production in CD4+ T cells was not due to an increased frequency of Th1 cells as neither the frequency of IFN-γ+ CD4+ T cells following PMA stimulation nor T-bet expression were increased in STAT4 risk allele carriers.
STAT4 risk individuals had a numerical increase in their plasma concentrations of IFN-γ, but there was a large interindividual variation and the difference was not statistically significant. This finding is perhaps not unexpected given that several factors may influence the IFN-γ production in vivo and that the observed effect of the risk allele was both cell-type specific and context dependent.
In line with the previously proposed augmented IFNAR sensitivity,20 STAT4 risk allele carriers also displayed increased pSTAT1 in CD8+ naïve T cells in response to IFN-α.
Mechanistically, we found that STAT4 risk allele carriers had higher total levels of STAT4 in CD8+ and CD4+ T cells following PHA/IL-2 stimulation. Given the intronic nature of rs7574865, it is conceivable that the risk SNP may enhance STAT4 mRNA expression on PHA/IL-2 stimulation, possibly by altering a transcription factor binding site for a transcription factor preferentially expressed at certain stages of T-cell differentiation. The low numbers of cells available from the patients with SLE and the need to isolate subsets of CD8+ T cells to study mRNA expression have so far precluded studies on the mRNA level.
IFN-γ and IL-12 are closely connected to the pathogenesis of SLE. IFN-γ is a proinflammatory cytokine, central for activation of B cells and macrophages. In murine models of lupus, Ifng or Ifngr deficiency prevents kidney damage and ameliorates disease.33–35 In patients with SLE, elevated levels of IFN-γ and IFN-γ-induced chemokines precede the accrual of serum IFN-α activity, autoantibodies and clinical lupus disease by several years.36 Furthermore, mRNA expression of genes induced by both IFN-α and IFN-γ, but not IFN-α-specific genes, correlates with renal flares.37 In terms of IL-12, increased serum levels are associated with the presence and severity of lupus nephritis and glomerular IL-12-positive cells are detected in patients with severe lupus nephritis.32 Given our finding of increased IL-12-induced IFN-γ production in STAT4 risk individuals, it is thus tempting to speculate that the increased risk for lupus nephritis in STAT4 risk patients is driven by an increased production of IFN-γ. A recent phase 1b clinical trial of IFN-γ blockade in patients with SLE with lupus nephritis showed no effect on the proteinuria on the aggregate level of patients.38 However, proteinuria was normalised in some patients on the individual level. Consequently, it would be interesting to stratify patients based on their STAT4 genotypes in forthcoming studies.
In the context of aberrant regulation of the IL-12/IFN-γ axis, it is interesting to note that several SLE risk loci encode genes that map to this pathway, for example, IL12A,39 IL12B,40 41 IL12RB2,42 JAK2,43 TYK2 44 and IFNG.45 Recently a SNP in TYK2, which is protective for SLE, was associated with a decreased response to IL-12R and IFNAR stimulation,46 suggesting the possibility that genetic dysregulation of the IL-12R response may be a common feature of SLE risk SNPs in locus belonging to the IL-12 pathway. Previous studies have found an additive effect of STAT4 and IRF5 risk SNPs,4 20 and it is conceivable that a combination of several risk gene variants determines the final STAT4 response and contributes to the clinical outcome. However, in this study, we did not have power to investigate possible gene interactions.
The lack of correlation between the IL-12 response and current treatment, and the fact that STAT4 risk allele carriers have a more severe disease outcome,4 15–17 may suggest that current standard lupus therapies do not target the pathway affected in STAT4 risk patients with SLE. The IL-12R signals through JAK2 and TYK2, whereas the IFNGR signalling is mediated through JAK1 and JAK2. Consequently, we compared a JAK2 and a TYK2-selective inhibitor with the pan-JAKi tofacitinib for their ability and selectivity of blocking IL-12, IFN-γ and IFN-α-induced activation of cells. Of the three JAKis evaluated, the TYK2i displayed the highest selectivity for IL-12. Importantly, the TYK2i efficiently blocked IL-12-induced pSTAT4 and IFN-γ production in cells from patients with SLE carrying the STAT4 risk allele, and the JAK2i blocked IFN-γ-induced activation of SLE STAT4 risk cells. Since STAT4 risk patients also had an increased response to IFN-α, and both IFNAR and IL-12R signalling is mediated via TYK2, TYK2 inhibition could be a therapeutic option especially useful in patients carrying the STAT4 risk allele.
In conclusion, this study establishes a molecular association between the strong genetic SLE risk variant in STAT4, rs7574865[T], and an augmented responsiveness to IL-12, which results in increased IFN-γ production in T cells. Furthermore, we demonstrate the feasibility to target the IL-12/IFN-γ axis in patients with SLE carrying the STAT4 risk allele with JAKis. These findings may have implications for patient stratification in clinical trials and may in the future aid in the selection of drugs for patients carrying the STAT4 risk allele.
Acknowledgments
We thank Rezvan Kiani for collecting samples from patients, Andrei Alexsson for bioinformatics support, Lisbeth Fuxler and Cane Yaka for technical assistance, the DISSECT consortium for valuable input and all patients who contributed samples to this study. Genotyping was performed by the SNP&SEQ Technology Platform, which is part of Science for Life Laboratory and the National Genomics Infrastructure at Uppsala University, supported by the Swedish Research Council (VR-RFI) and the Knut and Alice Wallenberg Foundation.
References
Footnotes
Handling editor Josef S Smolen
Contributors NH, YTB, A-CS and LR designed the study; DL and SR collected clinical data; JM and MKN provided reagents; M-LE and DL were responsible for enrolling patients; NH and MJ performed the experiments and analysed data; NH, MJ, YTB and LR interpreted data; NH and LR drafted the manuscript and all authors critically reviewed and approved the final version of the manuscript.
Funding This work was supported by grants from the Swedish Research Council for Medicine and Health (LR D0283001 and A0258801, YTB), the Swedish Rheumatism Association (LR, NH), King Gustav V’s 80-year Foundation (LR, NH), the Knut and Alice Wallenberg foundation (A-CS and LR 2011.0073, YTB), an AstraZeneca Science for Life Laboratory research collaboration grant (LR), the Swedish Society of Medicine and the Ingegerd Johansson donation (LR), Erik, Karin and Gösta Selanders foundation (NH), the Norwegian Research Council (YTB) and the Center for Innovative Medicine (YTB).
Competing interests MKN and JM are employed by AstraZeneca.
Patient consent Obtained.
Ethics approval The study was approved by the Regional Ethics board in Uppsala No. 00/399, 2007/024 and 2016/155.
Provenance and peer review Not commissioned; externally peer reviewed.