Article Text

Abstract
Objective RANKL is mainly expressed by synovial fibroblasts and T cells within the joints of rheumatoid arthritis patients. The relative importance of RANKL expression by these cell types for the formation of bone erosions is unclear. We therefore aimed to quantify the contribution of RANKL by each cell type to osteoclast differentiation and bone destruction during inflammatory arthritis.
Methods RANKL was specifically deleted in T cells (Tnfsf11flox/Δ Lck-Cre), in collagen VI expressing cells including synovial fibroblasts (Tnfsf11flox/Δ Col6a1-Cre) and in collagen II expressing cells including articular chondrocytes (Tnfsf11flox/Δ Col2a1-Cre). Erosive disease was induced using the collagen antibody-induced arthritis (CAIA) and collagen-induced arthritis (CIA) models. Osteoclasts and cartilage degradation were assessed by histology and bone erosions were assessed by micro-CT.
Results The inflammatory joint score during CAIA was equivalent in all mice regardless of cell-targeted deletion of RANKL. Significant increases in osteoclast numbers and bone erosions were observed in both the Tnfsf11flox/Δ and the Tnfsf11flox/Δ Lck-Cre groups during CAIA; however, the Tnfsf11flox/Δ Col6a1-Cre mice showed significant protection against osteoclast formation and bone erosions. Similar results on osteoclast formation and bone erosions were obtained in CIA mice. The deletion of RANKL on any cell type did not prevent articular cartilage loss in either model of arthritis used.
Conclusions The expression of RANKL on synovial fibroblasts rather than T cells is predominantly responsible for the formation of osteoclasts and erosions during inflammatory arthritis. Synovial fibroblasts would be the best direct target in RANKL inhibition therapies.
- Cytokines
- Inflammation
- Rheumatoid Arthritis
- Synovitis
- T Cells
Statistics from Altmetric.com
Introduction
Osteoclasts have the unique ability to resorb bone. Receptor activator of nuclear factor κB ligand (RANKL) is a key mediator of osteoclast formation and the binding of RANKL to RANK on haematopoietic osteoclast precursors directly induces osteoclast development and bone resorption.1–4 RANKL function is competitively regulated by the soluble decoy receptor osteoprotegerin (OPG).5 During physiological bone remodelling, RANKL is provided by osteoblast-lineage cells including osteocytes,6 ,7 while osteoblasts and chondrocytes provide the major source of RANKL for bone development.7 The effect of RANKL on T cells6 ,8 ,9 and B cells9 appears to be of little consequence for physiological bone development, although the deletion of RANKL specifically on B cells, but not T cells, was slightly but significantly protective of ovariectomy-induced bone loss.9
Rheumatoid arthritis (RA) is a chronic, systemic inflammatory disorder that characteristically produces an inflammatory synovitis and progressively destroys the articular cartilage and bones of the joints. In the collagen antibody-induced arthritis (CAIA) model of RA, the direct administration of anti-collagen antibodies initiates joint-targeted inflammation and bone destruction and the onset of arthritis is independent of T or B cells.10–13 The collagen-induced arthritis (CIA) model is, however, dependent on collagen-specific CD4+ T cells that initiate the joint inflammatory processes.14 Studies using T cell-dependent and T cell-independent arthritis models have demonstrated that RANKL global knockout mice or mice treated with RANKL inhibitors were protected against the formation of bone erosions, yet the development of inflammation was equivalent compared to RANKL-sufficient mice.15–18
The RANKL-expressing cell types responsible for driving osteoclastogenesis in vivo have not yet been identified in rheumatology research. Within the joint tissues from RA patients, RANKL is highly expressed on synovial fibroblasts and T cells.16 ,19–21 Cultured synovial fibroblasts can promote osteoclastogenesis when stimulated with 1,25-dihydroxyvitamin D3 (1,25(OH)2D3) or interleukin (IL)-17.20–22 Similarly, certain subsets of in vitro activated T cells are capable of stimulating osteoclastogenesis under specific conditions.16 ,19 ,23 ,24 Among them, Foxp3+ T cell-derived Th17 cells have been demonstrated to be the most capable of supporting osteoclastogenesis.23 Although these Th17 cells indeed express RANKL and contribute to osteoclastogenesis directly, the effect on osteoclastogenesis has mainly been attributed to their ability to upregulate RANKL expression on synovial fibroblasts, due to the action of IL-17A secretion.23–25 Thus, the direct contribution of RANKL derived from T cells versus synovial fibroblasts to osteoclast differentiation in arthritic joints still remains to be determined in vivo.
Using mice with targeted deletion of RANKL on T cells compared to synovial fibroblasts, we have elucidated that, in both T cell-independent and T cell-dependent models of autoimmune arthritis, the expression of RANKL on T cells appears to be of minor significance compared to the expression of RANKL on synovial fibroblasts for the formation of osteoclasts and bone destructive erosions. Therefore, the targeting of synovial fibroblasts with anti-RANKL agents might be sufficient to hamper osteoclast activity and protect against bone destruction in RA.
Materials and methods
Animals
All mice had a C57Bl/6 (H-2b) genetic background, and were housed under specific pathogen-free conditions and fed standard rodent chow and water ad libitum. T cell-specific RANKL-deficient, Tnfsf11flox/Δ Lck-Cre mice have been previously described.6 To achieve deletion of RANKL in the joint tissues, the promoter-specific Cre mice were inter-crossed with Tnfsf11flox/Δ mice. Mice expressing the collagen VI promoter26 were used to delete RANKL from joint mesenchymal cells including synovial fibroblasts to generate Tnfsf11flox/Δ Col6a1-Cre mice. Mice expressing the collagen II promoter27 were used for the targeting of chondrocytes including articular chondrocytes to generate Tnfsf11flox/Δ Col2a1-Cre mice. Labelling of Col6a1-Cre expressing cells was achieved by inter-crossing Col6a1-Cre mice with CAG-CAT-EGFP transgenic mice.28 All animal experiments were performed with the approval of the Institutional Review Board of the University of Tokyo.
Induction of CAIA or CIA
Male mice between 8 and 12 weeks old were administered 5 mg of Arthritogenic-CIA 5-Clone Cocktail (Chondrex) by tail vein injection on day 0, followed by 50 µg/mouse LPS by intraperitoneal injection on day 3; hereafter referred to as CAIA. The mice were monitored every 1 or 2 days for signs of arthritis.
For CIA, male mice between 10 and 12 weeks old were immunised intradermally at the base of the tail and then re-challenged 3 weeks later with an emulsion consisting of 50 μL of chicken type II collagen (Sigma-Aldrich; 4 mg/mL) and 50 μL of complete Freund's adjuvant (Difco Laboratories) containing heat-killed Mycobacterium tuberculosis H37Ra (3.3 mg/mL). The mice were then monitored every 3 or 4 days for 21 days.
For the clinical assessment of arthritis in both CAIA and CIA, see the online supplementary text.
Histology
Knee joints were collected from euthanised CAIA mice at day 14 or CIA mice at day 21. Age and sex-matched naive mice were used as controls. The paws were fixed in 4% paraformaldehyde overnight and decalcified in Osteosoft (Merck) at 4°C, then paraffin-embedded and sectioned (9 µm). Haematoxylin (Mayer's) and eosin Y (H&E) staining was used to visualise inflammatory cell infiltration into the joints as per the standard protocol. Safranin O (Sigma-Aldrich) staining was used to assess cartilage damage. Tartrate-resistant acid phosphatase (TRAP) staining was used for analysis of osteoclasts with counterstaining in methyl green (Vector). TRAP-positive (TRAP+) osteoclasts at the synovial pannus—articular cartilage interface were counted using an optical microscope. For cartilage damage scoring, see the online supplementary text.
Frozen sections were prepared from the knee joints of 2-week-old Col6a1-Cre CAG-CAT-EGFP mice. Nuclei were stained with DAPI for 10 min.
Micro-CT acquisition
Three-dimensional micro-CT analysis was performed on the knee and calcaneus of arthritic mice and control mice. CT scanning was performed using a ScanXmate-A100S Scanner (Comscantechno). Three-dimensional microstructural image data were reconstructed, and structural indices were calculated using TRI/3D-BON software (RATOC).
Flow cytometry
In vitro generated CD4+ T cells were isolated and activated using plate-bound anti-CD3 and anti-CD28 antibodies. After 3 days the cells were assessed for surface RANKL expression.
Synovial membrane and synovial fluid samples were carefully harvested, using a dissecting microscope, from the knees and ankles of CAIA or control mice. The excised membranes were minced and digested in type II collagenase (1 mg/mL; Worthington) in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 20% FBS (Sigma-Aldrich) for 2 h and cultured after washing. Confluent cells were trypsinised and depleted of myeloid cells by sorting into CD90.2+CD11b− synovial fibroblasts using FACS Aria. The resulting fibroblasts were then re-cultured in temperature-sensitive tissue culture plates (RepCell) for 3 days with 1,25(OH)2D3 (10−8 M) and prostaglandin E2 (PGE2) (10−7 M). The cells were then harvested without enzymatic digestion in cold PBS for assessment of surface RANKL expression on CD90.2+CD11b− cells. All FACS analysis was performed using Canto II with Diva software (BD Biosciences).
Statistical analysis
Results were expressed as mean±SEM. Statistical differences were analysed using one-way ANOVA followed by Dunnett's post hoc test when applicable or unpaired Student’s t test. Correlations of the eroded volume of bone versus the CIA score were performed using a two-tailed, non-parametric Spearman test.
Results
Effect of cell-specific RANKL deletion on the development of inflammation in CAIA
T cell-specific RANKL-deficient, Tnfsf11flox/Δ Lck-Cre mice have been previously described.6 Deletion of RANKL was achieved in joint mesenchymal cells including synovial fibroblasts by the inter-crossing of Tnfsf11flox/Δ mice with Col6a1-Cre mice. Tnfsf11flox/Δ Col6a1-Cre, Tnfsf11flox/Δ Lck-Cre and Tnfsf11flox/Δ mice developed synchronised swelling of the fore and hind paws, accompanied by rapid weight loss within 4 days after administration of the collagen antibodies (figures 1A, B). Moderate inflammation was achieved in all CAIA groups (Tnfsf11flox/Δ Col6a1-Cre, Tnfsf11flox/Δ Lck-Cre and Tnfsf11flox/Δ) with the average maximum score being reached by day 8 and maintained until around day 11, after which the signs of inflammation steadily reduced (figure 1A). There was no significant difference in arthritis score or paw swelling among the three groups (figure 1A–D).

The severity of collagen antibody-induced arthritis (CAIA) inflammation is comparable in T cell or synovial fibroblast RANKL-deleted mice. CAIA groups: Tnfsf11flox/Δ (light grey line), Tnfsf11flox/Δ Lck-Cre (dark grey line) and Tnfsf11flox/Δ Col6a1-Cre (black line). Graphs show (A) arthritis score, (B) weight loss, (C) fore and hind paw thicknesses, and (D) images of hind paw swelling. Individual experiments were performed five times, n=5–6 per group.
Efficiency of RANKL deletion on T cells and synovial fibroblasts
Isolated naive CD4+ T cells were activated in vitro and analysed to confirm the selective deletion of RANKL protein on Tnfsf11flox/Δ Lck-Cre cells compared to Tnfsf11flox/Δ cells (figure 2A). We confirmed the selective deletion of RANKL in 1,25(OH)2D3- and PGE2-stimulated Tnfsf11flox/Δ Col6a1-Cre synovial fibroblasts by FACS analysis after harvesting the cells without enzymatic digestion or EDTA. RANKL protein expression on the CD90.2+CD11b− synovial fibroblasts was dramatically and specifically diminished on Tnfsf11flox/Δ Col6a1-Cre cells (figure 2B).

Efficient deletion of RANKL in T cells and synovial fibroblasts. (A) CD4+ T cells were activated in vitro. Dot plots show gating on CD4+ T cells. Histogram overlays of cell surface expression of RANKL on Tnfsf11flox/Δ (black) and Tnfsf11flox/Δ Lck-Cre (grey solid) cells compared to isotype control (CTRL) (red). (B) Collagen antibody-induced arthritis (CAIA)-derived synovial membranes were sub-cultured in the presence of 1,25(OH)2D3 and prostaglandin E2 (PGE2). Dot plots show gating on CD90.2+CD11b− fibroblast cells. Histogram overlays of cell surface expression of RANKL on control Tnfsf11flox/Δ and Tnfsf11flox/Δ Col6a1-Cre compared to isotype control. Bar charts show cell numbers in (C) synovial fluids and (D) synovial membranes harvested from joints of control Tnfsf11flox/Δ and CAIA-challenged Tnfsf11flox/Δ, Tnfsf11flox/Δ Lck-Cre and Tnfsf11flox/Δ Col6a1-Cre mice. Individual experiments were performed four times, n=4 per group. (E) Frozen sections of the knee joints of Tnfsf11flox/Δ CAG-CAT-EGFP and Tnfsf11flox/Δ Col6a1-Cre CAG-CAT-EGFP naive mice. Scale bars: 200 μm. Significance: *p<0.05; n.s., non-significant.
Effects of cell-specific RANKL deletion on the recruitment of T cells to the inflamed joints in CAIA
CAIA substantially increased cellular infiltration into the joint synovial fluid (figure 2C) and synovial membrane (figure 2D). Consistent with clinical score, there was no apparent effect of the deletion of RANKL, either from T cells or synovial fibroblasts, on the total numbers of CD3ε+ or CD4+ cells infiltrating into the synovial fluid (figure 2C) or synovial membrane (figure 2D).
No effect of cell-targeted RANKL deletion on bone development and homeostasis
Col6a1-Cre mice were crossed with CAG-CAT-EGFP reporter mice (Col6a1-Cre CAG-CAT-EGFP reporter), confirming the targeting of synoviocytes and articular chondrocytes as previously described26 (figure 2E). We also confirmed that the bones of 12-week-old Tnfsf11flox/Δ Lck-Cre mice had normal bone volume (BV), trabecular bone mineral density (BMD) and cortical BMD, as previously reported for younger mice6 ,7 (figure 3A, middle set and panels C, D and E, white bars.) Here we show for the first time that Tnfsf11flox/Δ Col6a1-Cre mice do not exhibit any growth retardation or bone abnormalities (figure 3A, right set). All mice exhibit normal percentage BV/trabecular volume (TV), trabecular BMD and cortical BMD at 12 weeks of age (figure 3C–E, white bars). These results suggest that homeostatic bone turnover or osteoclast formation in the steady state of adult mice is not influenced by RANKL expressed by T cells or Col6a1-expressing cells including synoviocytes and articular chondrocytes.

Deletion of RANKL on T cells or synovial fibroblasts did not affect bone development or homeostasis. Micro-CT cross-section and longitudinal sections of (A) control naive and (B) collagen antibody-induced arthritis (CAIA) mice. Scale bars: 1 mm. Bar charts show bone densitometry for (C) bone volume (BV)/trabecular volume (TV), (D) trabecular bone mineral density (BMD), and (E) cortical BMD analysis for control (white bars) and CAIA mice (black bars), n=3–5 per group. n.s., non-significant.
Absence of periarticular or systemic osteopenia in CAIA
CAIA did not substantially affect the BV of the distal tibia, trabecular BMD and cortical BMD of Tnfsf11flox/Δ mice, Tnfsf11flox/Δ Lck-Cre mice or Tnfsf11flox/Δ Col6a1-Cre mice compared to controls. These results suggest that the transient inflammation induced during CAIA was not sufficient to induce periarticular or systemic osteopenia (figure 3C–E, black bars).
Effects of cell-specific RANKL deletion on osteoclast differentiation and bone erosions in CAIA
Histological analysis revealed that all CAIA mice exhibited inflammatory pannus formation (figure 4A, top) which was absent in control Tnfsf11flox/Δ mice. TRAP+ osteoclasts were observed at the synovial pannus–joint interface in CAIA Tnfsf11flox/Δ mice, and were absent in control Tnfsf11flox/Δ mice. The deletion of RANKL on T cells in CAIA was not protective against the formation of TRAP+ osteoclasts (figure 4A, middle). Counting of TRAP+ cells also revealed no difference in osteoclast numbers between Tnfsf11flox/Δ Lck-Cre and Tnfsf11flox/Δ mice (figure 4B).

Synovial fibroblast but not T cell-targeted deletion of RANKL is protective against collagen antibody-induced arthritis (CAIA)-associated bone erosions. (A) Histological sections of the knee joints of control and CAIA groups. H&E (upper) and tartrate-resistant acid phosphatase (TRAP) staining at low magnification and higher magnification (boxed areas, middle). Safranin O staining (lower). Black scale bars: 200 μm. (B) TRAP+ cells at the joint–pannus interface counted per section and per joint (upper bar charts) and the cartilage damage score assessment (lower bar chart), n=5–6 per group. (C) Micro-CT reconstructions of erosions (red) on the calcaneus (upper) and femur (lower) of control and CAIA mice. White scale bars: 1 mm. (D) Bar charts show percentages of erosion parameters for control and CAIA groups, n=5–6 per group. CTRL, control. Significance: *p<0.05, **p<0.005, ***p<0.001, ****p<0.0001. n.s., non-significant.
However, the deletion of RANKL on synovial fibroblasts in Tnfsf11flox/Δ Col6a1-Cre mice resulted in a striking reduction in TRAP+ osteoclast formation (figure 4A, right panels), despite profuse inflammation and pannus proliferation which were comparable to that of Tnfsf11flox/Δ CAIA mice (figures 1A, D and 4A, top). The TRAP+ cells observed at the articular surface were significantly fewer in Tnfsf11flox/Δ Col6a1-Cre mice than in either Tnfsf11flox/Δ Lck-Cre or Tnfsf11flox/Δ CAIA mice (figure 4B, top).
Compared to naive Tnfsf11flox/Δ control mice, all CAIA groups exhibited significant articular cartilage damage and loss of proteoglycan content as visualised by loss of safranin O stain (figure 4A, bottom). Indeed, the deletion of RANKL on either T cells or synovial fibroblasts had no protective effects on the cartilage damage score among CAIA groups (figure 4B, bottom).
Micro-CT analysis of the ankle and knee joints revealed that the deletion of RANKL on T cells in Tnfsf11flox/Δ Lck-Cre mice was not protective against the formation of bone erosions in CAIA (figure 4C, D). In contrast, the absence of RANKL on synovial fibroblasts in Tnfsf11flox/Δ Col6a1-Cre mice resulted in protection against the formation of erosions; both the surface volume and area of erosions were significantly reduced as compared to Tnfsf11flox/Δ CAIA mice (figure 4D, left and middle). Accordingly, there was a significant reduction in the remaining volume of bone in both Tnfsf11flox/Δ Lck-Cre and Tnfsf11flox/Δ mice as compared to control Tnfsf11flox/Δ mice, whereas the remaining volume of bone in CAIA Tnfsf11flox/Δ Col6a1-Cre mice was not reduced compared to non-arthritic Tnfsf11flox/Δ controls (figure 4D, right).
RANKL on articular chondrocytes does not contribute to CAIA-associated osteoclast differentiation and bone erosions
To exclude the possibility that protection against erosions on Tnfsf11flox/Δ Col6a1-Cre mice was partly due to the deletion of RANKL expression within articular cartilage, we deleted RANKL in chondrocytes, including articular chondrocytes, by crossing Tnfsf11flox/Δ mice with mice expressing the Col2a1 promoter to generate Tnfsf11flox/Δ Col2a1-Cre mice. Synchronised onset of arthritis accompanied by rapid weight loss was observed in both the Tnfsf11flox/Δ and Tnfsf11flox/Δ Col2a1-Cre mice, with no differences observed amongst the two CAIA groups (figure 5A). Histological analysis revealed similar formation of TRAP+ osteoclasts in the joints of Tnfsf11flox/Δ Col2a1-Cre mice and Tnfsf11flox/Δ mice (figure 5B). Counting of TRAP+ cells also revealed no difference in osteoclast numbers between Tnfsf11flox/Δ Col2a1-Cre and Tnfsf11flox/Δ mice (figure 4B, left). The deletion of RANKL on articular chondrocytes had no protective effect against cartilage damage (figure 5C). Micro-CT analysis of the ankle joints revealed that the deletion of RANKL on articular chondrocytes in Tnfsf11flox/Δ Col2a1-Cre mice was not protective against the formation of bone erosions compared to Tnfsf11flox/Δ CAIA mice (figure 5D).

RANKL expression by articular chondrocytes does not contribute to collagen antibody-induced arthritis (CAIA)-associated bone erosions. (A) CAIA score and weight loss of Tnfsf11flox/Δ and Tnfsf11flox/Δ Col2a1-Cre mice. (B) Tartrate-resistant acid phosphatase (TRAP) staining of arthritic knee joints of Tnfsf11flox/Δ and Tnfsf11flox/Δ Col2a1-Cre mice. Black scale bars: 200 μm. (C) Counting of TRAP+ cells at the joint–pannus interface and the cartilage damage score assessment. (D) Micro-CT reconstructions of erosions (red) on the calcaneus of control and CAIA mice. White scale bars: 1 mm. Bar charts show percentages of erosion parameters for control (CTRL) and CAIA groups. Individual experiments were performed three times, n=5 per group. Significance: *p<0.05, **p<0.005, ****p<0.0001. n.s., non-significant.
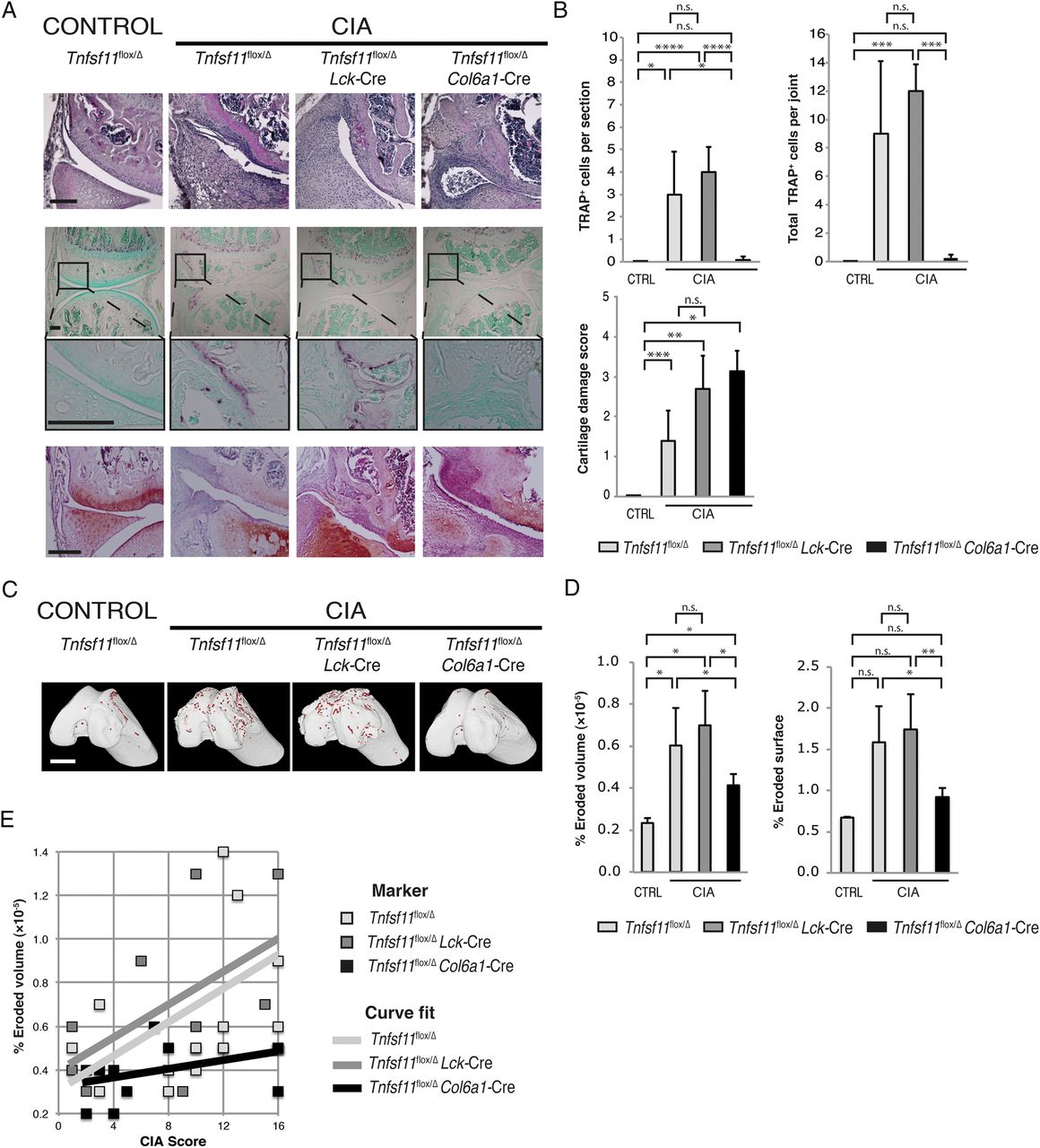
Synovial fibroblast but not T cell RANKL is required for CIA-induced osteoclast differentiation and bone erosions
Mice that developed clinical signs of CIA were histologically examined by H&E, TRAP and safranin O staining (figure 6A). The deletion of RANKL on T cells was not protective against the formation of TRAP+ osteoclasts (figure 6A, middle). Counting of TRAP+ cells revealed no difference in osteoclast numbers between Tnfsf11flox/Δ Lck-Cre and Tnfsf11flox/Δ mice (figure 6B) or bone erosions (figure 6C, D). However, the deletion of RANKL on synovial fibroblasts significantly diminished the appearance of TRAP+ osteoclasts in the joint (figure 6A, B), and reduced the volume and surface area of erosions as compared to either Tnfsf11flox/Δ or Tnfsf11flox/Δ Lck-Cre mice (figure 6D). The deletion of RANKL from either T cells or synovial fibroblasts was not protective against signs of cartilage damage in CIA mice, similar to the observation in CAIA mice (figure 6A, bottom, and B, lower). The CIA score was varied within each experimental group due to the C57BL/6 (H-2b) genetic background of the mice. Despite this variation, a positive correlation (shown as curve fit) between the CIA score and percentage of eroded volume was observed in Tnfsf11flox/Δ mice (p=0.027), but this correlation was lost in Tnfsf11flox/Δ Col6a1-Cre mice (figure 6E).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Formation of collagen-induced arthritis (CIA)-associated bone erosions are reduced when RANKL is deleted on synovial fibroblasts but not T cells. (A) Histological sections of the knees of control or CIA groups (inflammatory joint score of 4). H&E staining (upper) and tartrate-resistant acid phosphatase (TRAP) staining, lower and higher magnification (middle). Safranin O staining (lower). Black scale bars: 200 μm. (B) TRAP+ cells at the joint–pannus interface counted per section and per joint (upper bar charts) and the cartilage damage score assessment (lower bar chart), n=4 per group. (C) Micro-CT reconstructions of erosions (red) on the femur of control and CIA mice. White scale bars: 1 mm. (D) Bar charts show percentages of erosion parameters for control and CIA groups. (E) Chart of percentages of eroded volume of bone plotted against CIA score with curve fit for each experimental group, n=10–14 per group. CTRL, control. Significance: *p<0.05, **p<0.005, ***p<0.001, ****p<0.0001. n.s., non-significant.
Discussion
T cells play an important role in the onset and pathogenesis of autoimmune arthritis, in particular in the initial phase of autoimmune reactions and inducing local inflammation in the joints.14 However, there has been controversy over the role of T cells in the bone destruction phase, since T cells are not typically activated in the human RA synovium29 and T cells or T cell cytokines are not required for bone destruction in certain models of arthritis.14 The role of osteoclasts in arthritic bone erosion has been established, but it has been unclear whether T cells directly contribute to the enhanced osteoclastogenesis. Both fibroblasts and T cells can be induced to express RANKL and support osteoclastogenesis in vitro; however, as it remains technically challenging to establish the relative expression levels of RANKL on fibroblasts or T cell subsets in the joint, the source of RANKL for osteoclastogenesis in vivo has remained elusive.
In order to address the question of the direct role of RANKL expression by T cells on osteoclastogenesis, we deleted RANKL on T cells using Tnfsf11flox/Δ Lck-Cre mice, and revealed that this was not protective against the formation of osteoclasts or erosions in inflamed joints. It is theoretically possible that certain CD4+ T cells may not express Lck, but since we were able to demonstrate that RANKL was almost completely eliminated on the in vitro stimulated CD4+ T cells from Tnfsf11flox/Δ Lck-Cre mice, we presume that all T cell subsets were adequately targeted. The deletion of RANKL on synovial fibroblasts using Tnfsf11flox/Δ Col6a1-Cre mice was significantly protective against osteoclast formation and bone erosions. We also took advantage of the Col2a1-Cre line to confirm that RANKL expression by articular chondrocytes did not contribute to the bone erosions induced during inflammatory arthritis. We have furthermore verified that deletion of RANKL expression on these cell types has no effect on cartilage destruction.30 Thus we have provided in vivo genetic evidence that synovial fibroblasts rather than T cells are responsible for bone erosion, although we cannot rule out the possibility that these results were influenced by unexpected Cre expression in non-target cells.
RANKL also plays a role in the regulation of immune responses. RANKL is crucial for the differentiation of medullary thymic epithelial cells, required for deleting autoreactive T cells,31–34 and the development of lymph nodes.35 RANKL promotes dendritic cell survival and cytokine production36–40 and is involved in Treg cell generation.41 ,42 However, T cell-dependent and T cell-independent arthritis studies have not reported any significant changes in immune responses.15–18 Consistent with this, we observed no significant difference in the level of inflammation in CAIA. We utilised CIA in order to assess the effect of the cellular source of RANKL on the degree of bone erosions in a T cell-dependent arthritis model. However, we observed a degree of variability in the CIA-induced inflammation due to the use of C57BL/6 (H-2b) mice. Therefore, we propose that, in order to more precisely study the effect of RANKL on T cells in immune responses and osteoporosis, it is necessary to perform the experiments in DBA/1J mice, which are more susceptible to CIA.
These results identify that synovial fibroblasts are the principle RANKL-expressing cells and are responsible for the formation of bone destructive osteoclasts during inflammatory arthritis and show that the targeting of RANKL specifically on synovial fibroblasts could be effective in preventing the debilitating consequences of bone destruction in RA.
Acknowledgments
Mice expressing the collagen II promoter were kindly gifted from Dr T Kanemoto (Osaka Bisocience Institute) with permission from Professor R Behringer. CAG-CAT-EGFP transgenic mice were kindly gifted from Professor J Miyazaki (Osaka University). The authors would like to thank K Okamoto, T Negishi-Koga, M Hayashi, M Shinohara, M Oh-hora, H Takaba, A Terashima, K Kusubata, A Suematsu, K Nagashima, S Fukuse, T Ando and C Tsunoda for their discussions and assistance.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
Handling editor Tore K Kvien
Contributors LD performed the experiments, interpreted the results, and wrote the manuscript. NK, MMG, SS and TN shared experimental expertise, helped with data interpretation and edited the manuscript. MA and GK bred the Col6a1-Cre mice and edited the manuscript. TN and HT conceptually designed the project. HT carried out critical and conceptual editing of the manuscript.
Funding This work was funded in part by a grant for the ERATO Takayanagi Osteonetwork Project from JST; a Grant–in-Aid for Challenging Exploratory Research from the Japan Society for the Promotion of Science (JSPS) (25670636); a Grant-in-Aid for Specially Promoted Research from the JSPS (15H05703). MMG and LD were supported in part by JSPS Postdoctoral Fellowships for Overseas Researchers (2200131 and 2301114 respectively). NK was supported in part by JSPS Research Fellowships for Young Scientists (224341) and a Grant-in-Aid for JSPS Fellows and a Grant-in-Aid for Young Scientists A from the JSPS (26713015). GK and MA were supported in part by IMI-funded project BeTheCure (115142-2) and GSRT national project INNATE FIBROBLAST (ERC06, co-financed by ESF and NSRF 2007-2013). The authors thank the Infrafrontier-GR infrastructure (co-financed by ERDF) for animal housing.
Competing interests None declared.
Ethics approval All animal experiments were performed with the approval of the Institutional Review Board of The University of Tokyo. This study does not include human samples/subjects.
Provenance and peer review Not commissioned; externally peer reviewed.