Article Text

Abstract
Objective To identify shared and differential molecular pathways involved in the pathogenesis of atherosclerosis (AT) and cardiovascular disease (CVD) in systemic lupus erythematosus (SLE), primary antiphospholipid syndrome (APS) and APS associated with SLE (APS plus SLE).
Methods 129 patients (42 APS, 31 APS plus SLE and 56 SLE) and 61 healthy donors were included. Microarray expression profiling was performed in monocytes. RT-PCR of selected genes and western blot were used to validate microarray data. Clinical and inflammatory parameters were also analysed.
Results Compared with controls, 555, 1224 and 518 genes were differentially expressed in monocytes from SLE, APS plus SLE and APS patients, respectively. Approximately 25–30% of differentially expressed genes were related to AT and CVD. Each disease displayed a specific AT/CVD/Inflammation-related gene signature. Compared with SLE, APS showed alterations in mitochondria biogenesis and function and oxidative stress. Besides the interferon signature, found in APS plus SLE and SLE patients, various genes mediating atherosclerotic/inflammatory signalling were also differentially expressed in APS plus SLE. IgG-anticardiolipin (aCL) titres independently predicted both atherosclerotic and thrombosis in APS plus SLE. Moreover, a significant correlation of IgG-aCL titres with mRNA levels of certain inflammatory molecules in monocytes was further noticed. In vitro treatment of monocytes with IgG-aCL promoted an increase in the expression of the genes most significantly changed in APS plus SLE versus healthy donors.
Conclusions Gene expression profiling allows the segregation of APS, APS plus SLE and SLE, with specific signatures explaining the pro-atherosclerotic and pro-thrombotic alterations in these highly related autoimmune diseases.
- Systemic lupus erythematosus
- atherosclerosis
- cardiovascular disease
- gene profile
Statistics from Altmetric.com
Introduction
The development of atherosclerosis and cardiovascular disease (CVD) in antiphospholipid syndrome (APS) and systemic lupus erythematosus (SLE) involves genetic factors as well as other acquired and modifiable risk factors (eg, hypercholesterolaemia, diabetes mellitus and hypertension). Inflammatory components of the immune response, as well as autoimmune elements (eg, autoantibodies, autoantigens and autoreactive lymphocytes), seem to be also involved in these processes. In addition, oxidative stress as well as dyslipidemia and various systemic inflammation mediators, including cytokines, chemokines, prothrombotic molecules and adhesion receptors, among others, have been implicated in the development of these vascular pathologies.1 ,2 APS and SLE share several clinical and molecular features, but also have some unique distinguishing characteristics. Thus, antiphospholipid antibodies (aPLs) and other autoantibodies are responsible for the development of atherothrombosis in APS. aPLs promote the overexpression of tissue factor (TF) and protease activated receptors (PARs).3–5 They are also responsible for the altered protein profile of monocytes and related to thrombosis development including overexpression of annexins I and II or RhoA proteins among others.6 Besides, aPLs promote oxidative perturbations and mitochondrial dysfunction7–9 and also trigger an inflammatory cascade with increased expression of several cytokines, chemokines and mediators of endothelial dysfunction.10 ,11 Furthermore, aPLs cross-react with oxidised low-density lipoproteins (ox-LDLs), thus accelerating their influx into macrophages and promoting monocyte activation and atherosclerosis development.12 Moreover, serum levels of anticardiolipin (aCL) antibodies correlate with the incidence and severity of acute coronary syndrome, myocardial infarction and stroke.13 ,14
Relevant factors directly influencing the development of atherosclerosis and CVD in SLE comprise drug therapy, immune complex generation and changes in innate immune responses, complement activation, endothelial dysfunction, oxidative stress, increased production of adipokines, dysfunctional lipids and changes in the production and activity of a complex network of cytokines.15 ,16
APS associated with SLE (APS plus SLE) is diagnosed in patients having polyautoimmunity, that is, who have SLE and have also suffered thrombotic events or adverse pregnancy outcomes in the presence of aPLs.17 Although APS plus SLE and APS patients have similar clinical profiles, heart valve disease, haemolytic anaemia, low C4 levels and neutropenia seem to be more common in patients with APS plus SLE.18 All that specific characteristics of those three pathologies have significant implications for the therapy. Therefore, it is of great relevance to clearly define each clinical entity and evaluate the patient based upon a complete knowledge of the underlying disease processes.
The pathogenesis of atherosclerosis involves various cell types of the immune system, having been shown that monocytes play an essential role. In addition to modulate lipid metabolism, monocytes secrete inflammatory cytokines, chemokines and reactive oxygen species that drive the pathogenesis. They also produce TF and proteases that contribute to thrombosis and atherosclerotic plaque rupture.19 ,20 Characterising the monocytes molecular signature may provide an alternative and effective way to identify heterogeneous autoimmune patient's subpopulations for targeted therapies. Microarray analysis is a broad-based profiling method that permits the concomitant comparison of gene expression profiles among different study groups, revealing active networks of interrelated genes within subpopulations under study.21
Few microarray studies in autoimmunity have been reported, and only some published reports have described results obtained using microarray analysis of peripheral blood mononuclear cells (PBMC) populations from patients with SLE.22–25 Yet, none of them have performed comparative studies in purified monocytes from APS, APS plus SLE and SLE.
Thus, by using microarray technology in combination with other protein analyses, the present study intended to identify shared and differential molecular pathways involved in the pathogenesis of atherosclerosis and CVD in these autoimmune disorders.
Patients and methods
In total, 126 patients, 41 with APS, 31 with APS plus SLE and 54 with SLE, as well as 61 healthy donors were included in the study (during a period of 24 months) after ethics committee approval was obtained. Subjects were selected among patients with stable disease for more than 6 months, without infections, abortions, thrombosis or changes in their treatment protocol. All patients provided written informed consent. None of the healthy controls had a history of autoimmune disease, bleeding disorders, thrombosis or pregnancy loss. The characteristics of the patients and the controls are shown in table 1.
Clinical and laboratory parameters for APS, APS+SLE and SLE and controls cohorts at enrolment
Blood samples
Plasma and serum samples, and purified monocytes (non-monocytes depleting kit, Miltenyi Biotech, Bergisch Galdbach, Germany), were obtained from peripheral venous blood samples as described elsewhere (see online supplementary methods).26 ,27
Flow cytometry analyses, Flow-cytomix and analysis of oxidative stress biomarkers in white blood cells and plasma
(See online supplementary methods for details).
B-mode ultrasound IMT measurements and thrombosis assessment
B-mode ultrasound imaging for carotid intimate media thickness (CIMT) measurements was performed as previously described28 ,29 by using a Toshiba equipment (Aplio platform) equipped with 7–10 MHz broadband linear array transducers. For further details, see supplementary methods.
Microarray analysis
Microarray studies were performed in an Agilent G4112F platform (Whole Human Genome Microarray 44k) using the One-Color gene expression system. These microarrays contain ∼41 000 human genes and transcripts with one 60-mer oligonucleotide probe representing each sequence. See supplementary methods for further details. The raw microarray data were deposited in the Gene Expression Omnibus database of the National Center for Biotechnology Information (accession no. GSE50395).
Quantitative real-time PCR
Changes of a number of transcripts highlighted by array results or related to the inflammatory/oxidative processes were validated by quantitative real-time RT-PCR using the LightCycler thermal cycler system (Roche Diagnostics, Indianapolis, USA), using GAPDH as housekeeping gene, as described elsewhere.26 ,27
Western blot
Specific antibodies against IL6R, SLC25A27 (Abnova, Heidelberg, Germany), IFIT1 (Abcam, Cambridge, UK) and GPx8 (Santa Cruz, Madrid, Spain) were used to determine protein levels by western blotting.26 ,27
Purification of IgG and in vitro exposure of normal monocytes to aPL antibodies
IgG from the pooled sera of seven patients with APS plus SLE (characterised by high titres of aCL antibodies, ie, >120 IgG phospholipid units) and from the pooled sera of seven healthy subjects (as controls) was purified by protein G-Sepharose high-affinity chromatography (MAbTrap kit; Amersham Biosciences). Purified normal monocytes (1.5×106 cells/mL) were incubated either with normal human serum (NHS)–IgG (500 µg/mL) or purified APS plus SLE patient-IgG (500 µg/mL) for 6 h at 37°C.
Statistical analysis
All data were expressed as mean±SD. Statistical analyses were performed with SSPS V.15.0 (SPSS Inc., Chicago, Illinois, USA). Following normality and equality of variance tests, comparisons were made by paired Student t test or alternatively by a non-parametric test (Mann–Whitney rank sum test). Correlations were assessed by Pearson product–moment correlation, and association studies were performed through χ2 test. The independent association between different variables in univariate analysis was determined by multivariate regression analysis. Differences were considered significant at p<0.05.
Results
Microarray study: functional categorisation of genes differentially expressed among APS, APS plus SLE and SLE patients in the area of atherosclerosis, inflammation and CVD
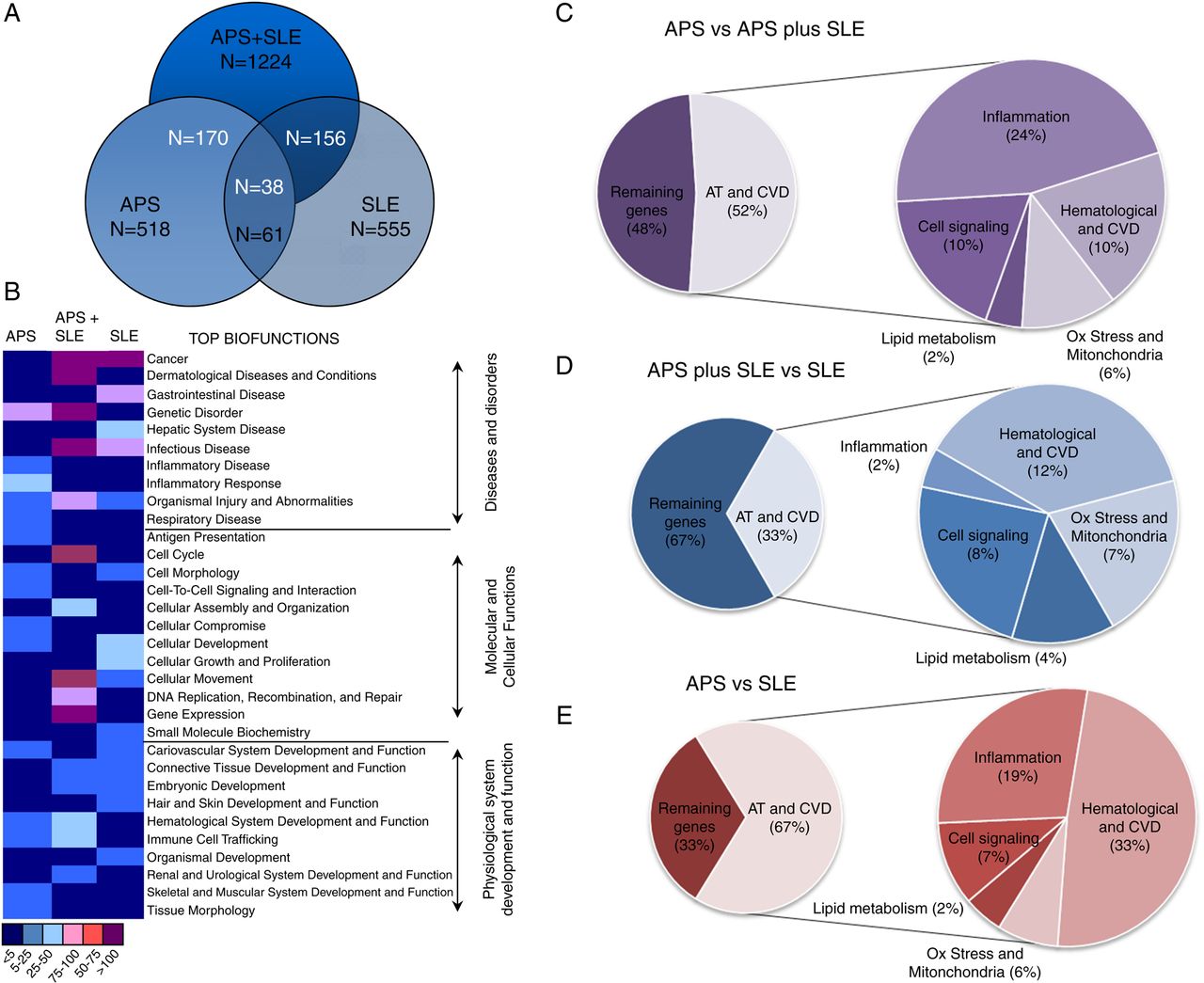
The number of genes differentially expressed in monocytes from patients versus controls was found significantly higher in APS plus SLE (1224) in relation to APS (518) and lupus patients (555). A comparative analysis allowed us to identify 1243 genes differentially expressed between APS and SLE, and 605 genes between APS plus SLE and SLE. Interestingly, only 220 genes were found differentially expressed in monocytes from APS patients compared with those of APS plus SLE, indicating a relatively small change between these two pathologies (figure 1A).

Overlapping and functional categorisation of gene expression signatures among antiphospholipid syndrome (APS), APS plus systemic lupus erythematosus (SLE) and SLE patients. (A) Venn diagram illustrating the overlap between the gene signatures that distinguish APS, APS plus SLE and SLE. Numbers in the diagram refer to number of genes. Those inside the circles refer to the number of genes differentially expressed in each respective disease versus healthy donors. The numbers inside the crossing circles refer to the genes simultaneously expressed in two or the three diseases. (B) Heat map of deviated functional categories in differentially expressed genes of APS, APS plus SLE and SLE patients versus healthy donors. Each category (distributed by top biofunctions) is ranked by a range of colours (purple, pink, light blue, dark blue) showing the number of genes differentially expressed. (C–E) Diagrams showing the functional categorisation of genes differentially expressed among diseases in the area of atherosclerosis and cardiovascular disease (CVD). Differentially expressed genes were classified and used for computational analysis to identify potential functional pathways and networks using the Ingenuity Pathways Analysis Knowledge Base (Ingenuity Systems).
Molecular network analysis showed that 20–30% of the total number of genes differentially expressed in the three pathologies was directly or indirectly related to atherosclerosis, inflammation and CVD. As a general feature, the main alterations were derived from the change in the expression of specific genes for each pathology, even though the number of genes differentially expressed was similar, and all of them belonged to the gene cluster of atherosclerosis, inflammation and CVD (figure 1B–E, see online supplementary figures S1 and S2).
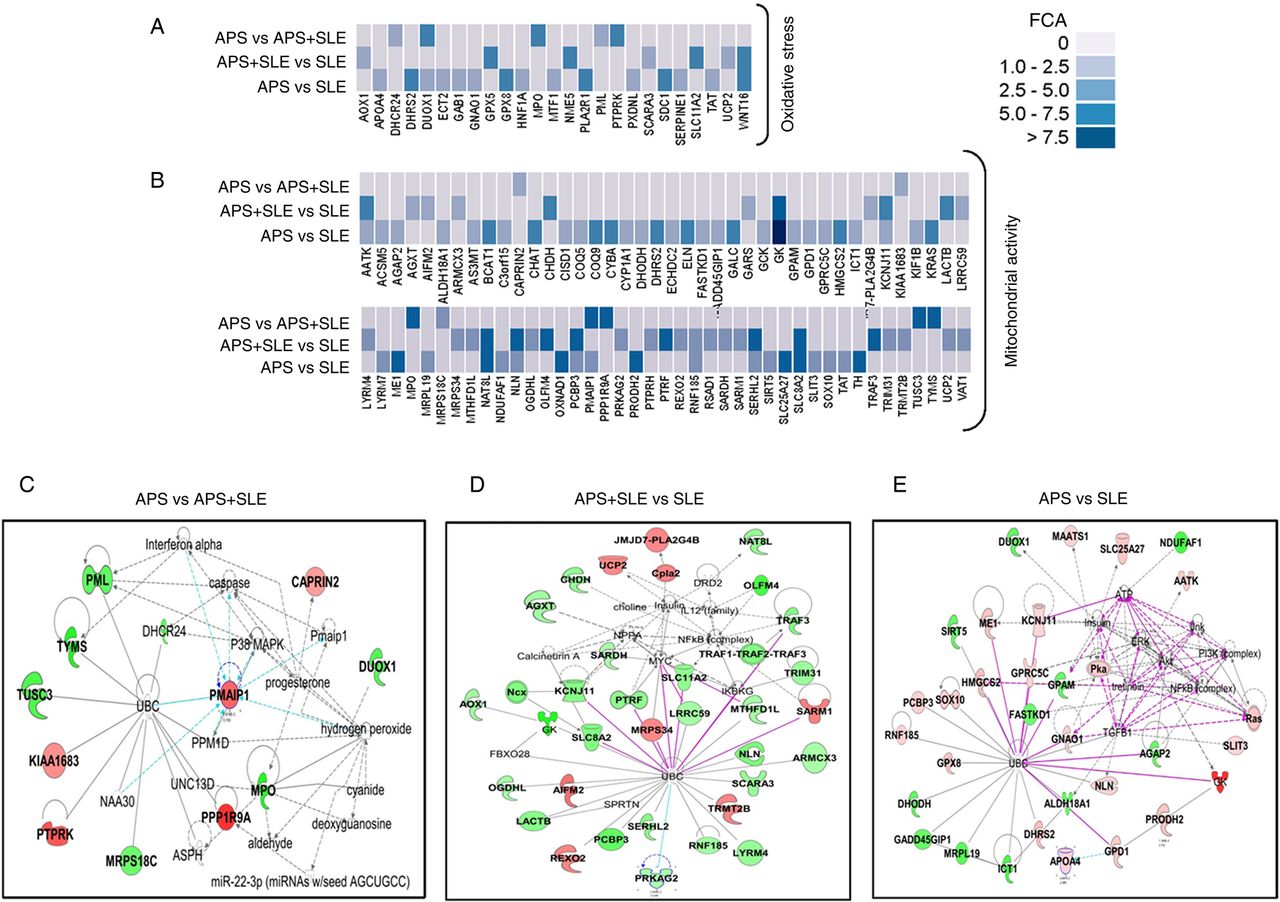
In relation to SLE monocytes from APS patients displayed differentially expressed genes involved in the biogenesis and function of mitochondria, including small molecule transporters (SLC25A27), regulators of membrane polarisation and potential (TP53TG5). Also, some genes related to oxidative stress and antioxidant defence were found differentially expressed, with increased expression of GPx8, CYBA and PXDN. Many other genes related to atherosclerosis were found upregulated, including those codifying for vascular endothelial growth factor receptors (KDR, FLT4), blood coagulation and circulation (ITGA2, ELN, LPL), vascular endothelial growth factor (VEGF) signalling (PI3 kinase signalling (PIK3R3, PIK3R5), small G-protein signalling (KRAS, SHC2), phospholipase A2 (PLA2G2E, PLA2G5) and protein phosphatases (PPP3R2)), cell adhesion molecules (CDH5), extracellular protease inhibitors (SERPINE1, PLG, TIMP3), transcription factors (ID3), chemokines (CXCL5), lipoprotein signalling and cholesterol metabolism (APOA4, CEL, HMGCS2, PRKAG2, APOF) and fatty acid metabolism (ACOXL, ACSM5, GK, GK5, GPD1, LPL). In the inflammatory pathway, a number of genes were found differentially expressed including chemokines, interleukins and their receptors, interferons, growth factors, genes belonging to the tumour necrosis factor (TNF) super family, interferon (IFN) responsive genes and genes regulators of the inflammatory response (figures 2 and 3). The most relevant group of genes that mediate inflammatory signalling in cells included those related to the Wnt signalling pathway, the NF-κB signalling pathway and the mitogen-activated protein kinase (MAPK) signalling pathway.
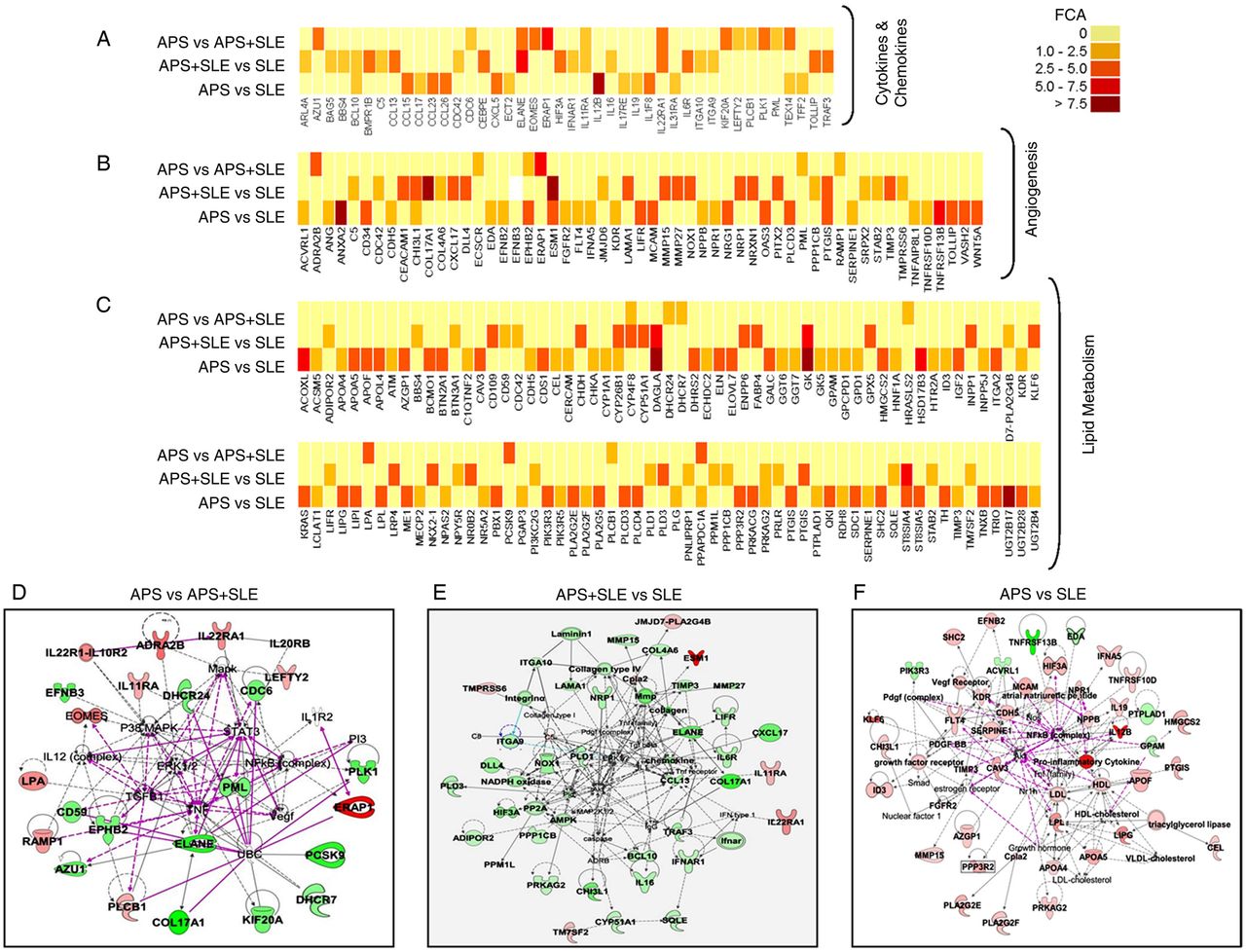
Functional categorisation of genes differentially expressed between monocytes of antiphospholipid syndrome (APS), APS plus SLE and systemic lupus erythematosus (SLE) patients in the area of atherosclerosis, inflammation and cardiovascular disease. (A–C) Heat maps depicting the expression microarray data for the genes differentially expressed between the three autoimmune conditions with p<0.05. FCA denotes fold change absolute. (D–F) Gene networks showing inter-relationship between differentially expressed genes in the three disorders using Ingenuity Pathways Analysis software. Overexpressed genes are shown in red and under-expressed genes appear in green. Direct interactions appear in the network diagram as a solid line, whereas indirect interactions as a dashed line.

Functional categorisation of genes differentially expressed between monocytes of antiphospholipid syndrome (APS) and systemic lupus erythematosus (SLE) patients in the area of oxidative stress and mitochondrial activity. (A, B) Heat maps depicting the expression microarray data for the genes differentially expressed between the three autoimmune conditions with p<0.05. FCA denotes fold change absolute. (C–E) Gene networks showing inter-relationship between differentially expressed genes in the three disorders using Ingenuity Pathways Analysis software. Overexpressed genes are shown in red and underexpressed genes appear in green. Direct interactions appear in the network diagram as a solid line, whereas indirect interactions as a dashed line.
In addition to the interferon signature (represented by genes such as IFI27, IFI35, IFI44, IFI44L, IFI6, IFIT1, IFIT5, IFITM1, IFITM4P, etc.), shared by APS plus SLE and SLE patients, differentially expressed atherosclerotic/inflammatory genes further included in APS plus SLE a significant number of those codifying for cytokines (IL6, IL11RA, IL16, IL22RA, IFNAR1) chemokines and their ligands/receptors (CCL13, CXCL17), adhesion molecules (COL4A6, NRP1) and genes mediating inflammatory response (ITGA10, TOLLIP, NOX1, CYP26B1, LIFR, NLRP11, TRAF3, etc.) (figures 2 and 3).
Monocytes from APS and APS plus SLE patients shared a high number of altered genes, with the most relevant differences found in those codifying in APS for some atherogenic molecules such as lipoprotein a (LpA), a recognised independent risk factor for premature atherosclerotic coronary heart disease,29 and cytokine receptors such as IL22RA1, which is produced by PBMCs cultured in the presence of aPLs, suggesting its role in the pathogenesis of APS30 (figures 2 and 3).
To further validate the data obtained, we performed two set of analysis:
Measurement of protein production by monocytes (see online supplementary figure S3), which suggested that the changed expression of some genes resulted in altered protein levels, thus providing evidence for a pathogenic role in the three studied diseases.
Monocytes from a number of patients of each pathology included in the study (5 APS, 7 APS+SLE and 10 SLE, and 10 controls) were evaluated again 2 years after the first blood sample collection to analyse the stability of changes observed in the array. For this purpose, we performed RT-PCR of various genes found changed in the microarray and validated in the first study. Results (see online supplementary figure S4) demonstrate that gene expression in the second sample collection changed in the same way when they were first analysed. Thus, our data support the theory that there is a specific Ath/CVD/inflammation signature characteristic of each disease, which remains stable along time.
Relationship between clinical and molecular markers of atherothrombosis and the presence of antiphospholipid antibodies in APS plus SLE patients
Coexistence of APS and the presence of aPLs are recognised risk factors for subclinical atherosclerosis and CVD development in lupus. Thus, we further searched for a link between clinical and molecular markers of atherosclerosis, inflammation and CVD, and the presence of aPLs.
First, we found that, contrary to APS, APS plus SLE and SLE patients displayed a significant number of inflammatory deregulated markers (table 2). Yet, approximately 35% of APS plus SLE patients showed increased pathologic IMT, while in LES the frequency was 20%. Multivariate analysis showed that aCL titres independently predicted the occurrence of thrombosis (standardised ß coefficient 0.463; p=0.001) and the presence of an increased CIMT (standardised ß coefficient 0.318; p=0.012), thus supporting an atherogenic/prothrombotic role for aCL antibodies of IgG isotype in APS plus SLE (figure 4A,B).
Parameters related to inflammation and oxidative stress in APS, APS+SLE and SLE patients

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Association and correlation studies and in vitro effects of IgG-APS+SLE on the expression of the most significantly differentially expressed genes in antiphospholipid syndrome (APS)+SLE in the area of atherosclerosis and cardiovascular disease (CVD). (A,B) Relationship between IgG isotype anticardiolipin antibodies levels and the presence of an increased carotid intimate media thickness (CIMT) and (A) or the occurrence of thrombotic events (B). * indicates significant differences either versus patients without increased CIMT or versus patients without thrombosis (p<0.05). (C–H) Correlation analyses between IgG isotype anticardiolipin antibodies levels and mRNA relative expression levels of vascular endothelial growth factor, IL-8, IL-1, MCP-1 (C–F), plasma levels of tPA (G), and GPx activity (H). (I) Monocytes isolated from healthy donors were incubated for 6 h with IgG-APS+SLE or IgG-normal human serum (NHS). Then, total RNA was extracted and RT-PCR of selected genes was performed as described in ‘Materials and methods’. Values are means and SEM from four independent experiments. *Significant differences (at p<0.05) versus monocytes treated with IgG-NHS.
Plasma, cell surface and mRNA analyses of inflammatory and oxidative stress markers validated and complemented some of the results obtained in the array in APS plus SLE (table 2). We further found a significant correlation between IgG-aCL titres and monocytes mRNA levels of some well-known inflammatory molecules, including VEGF, IL-8, IL-1 and monocyte chemoattractant protein-1 (MCP-1), endothelial dysfunction markers (tPA), as well as antioxidant defence markers such as the glutathione peroxidase activity (figure 4C–H).
Moreover, in vitro treatment of monocytes with IgG-aCL significantly changed the expression of the genes involved in those processes, including some of those found most significantly altered in their expression in APS plus SLE by using the microarray approach versus healthy donors, such as CCL2, IL11RA, scl25a27, ARHGEF5, IFIT1 and PPARG (figure 4I).
Discussion
This is the first study drawing simultaneously gene expression patterns of monocytes in patients with APS, APS plus SLE and SLE. A number of studies have compared gene expression in PBMC from patients with SLE versus healthy individuals or other autoimmune conditions,30–35 but only a few studies have focused on gene expression of cell sub-populations pertinent to SLE pathogenesis.36–39 From those studies, we could conclude that while array analysis of PBMC provide some useful information, the use of purified cell subsets identifies many more differentially expressed disease-specific genes. Such analyses provide substantial advantages in the search for diagnostic and prognostic biomarkers in autoimmune diseases. In the present study, the comparative gene profiling of three highly related autoimmune disorders identified multiple previously unreported genes differentiating them, thus enhancing our current understanding of these three diseases in aspects related to atherosclerosis and CVD.
APS, APS plus SLE and SLE have been associated with accelerated atherosclerosis or other types of vasculopathies leading to increased cardiovascular and cerebrovascular disease risk. Traditional risk factors, as well as systemic inflammation including cytokines, chemokines, adipokines, proteases, autoantibodies, adhesion receptors, oxidative stress biomarkers and a plethora of intracellular signalling molecules, have been implicated in the development of these vascular pathologies.1 ,2 ,40 Yet, the characteristics of vasculopathies may significantly differ depending on the underlying disease.
Our microarray analysis revealed that there are a significant proportion of genes differentially expressed in these three pathologies that codify for proteins that may be involved in the development of atherosclerosis and CVD. Moreover, by integrating the gene profiling data from monocytes of APS, APS plus SLE and SLE patients in the areas of CVD and oxidative stress using bioinformatics tools, disease-associated gene networks were identified. Pathways involved in cellular growth, thrombosis, inflammation (ERK1/2, AKT, PI3K, MAPK, p38MAPK) and immune reactions (STAT3, NFKB) were further recognised, implying an important role in the pathogenesis of the three autoimmune disorders.
Comparing among the three diseases, the high specificity found in the genes differentially expressed on each pathology would explain the differences and incidence of clinical manifestations. Thus, compared with SLE patients, a specific feature of APS monocytes was a significant number of differentially expressed genes involved in the mitochondria biogenesis and function, oxidative stress and antioxidant defence. These alterations have been previously reported to be directly related to thrombosis development in APS patients.8 ,9 A strong interferon signature was found in monocytes from SLE and APS plus SLE patients, thus corroborating previous studies that identified upregulation of type I IFN response genes in SLE22 ,25 ,41 and explaining the concordance of some clinical features between both disorders. In addition, a number of genes were differentially expressed in APS plus SLE that might further account for the pro-atherothrombotic tendency of these patients. Finally, a close relationship between APS and APS plus SLE patients was found in the group of genes belonging to the cluster of atherothrombosis, probably as a consequence of the aPL presence, which seems to drive this pathology in both autoimmune conditions.26 ,42 ,43
A recent study demonstrated a direct relationship between the presence of dysfunctional high-density lipoprotein (HDL) and of carotid plaque in SLE patients and the alteration of monocyte atherosclerosis-related transcripts, such as CCL2, TNFα and PDGFRß.44 Accordingly, our array also showed increased expression of PDGFRß in APS patients and PDGFRL in SLE patients. No changes were demonstrated in gene expression of CCL2 and TNFα, but some genes of the TNF family (TNFRSF17 in SLE and TNFRSF1A in APS) were found increased compared with control group.
Our study further suggested that APS plus SLE patients not only display a significantly different gene profile but also appear to be at greater risk of developing certain pathological features compared with those SLE patients who do not have aPL. Our results indicate that aPL-positive lupus patients have an increase in both markers of early atherosclerosis development and thrombotic events. A question that arises from that data is whether certain vascular manifestations, usually ascribed to lupus, are seen mainly or exclusively in those who have aPL.
Previous studies have shown that in APS plus SLE patients some clinical features that might be consistent with APS may not be caused by the presence of aPL.17 Indeed, these manifestations may be caused by some other aspects of lupus. Nevertheless, our present data further indicate a relevant influence of aPL on SLE-specific thrombosis development, as demonstrated by the relationship between IgG-aPL titres in APS plus SLE patients and the occurrence of thrombotic events. That influence was further supported by the correlation between aCL-IgG titres and the levels of monocytes mRNAs codifying for proteins directly related to thrombosis development.
Lupus patients have a 50-fold higher risk of developing atherosclerosis beyond the conventional risk factors.45 In primary APS, IgG aPL has been demonstrated to be an independent predictor of increased IMT, a marker of atherosclerotic vascular disease.9 ,46 We provide further evidence that aPL may be an additive risk for atherosclerosis in SLE that is not accounted for by the traditional risk factors (ie, the significant association found between IgG-aPL titres and premature atherosclerosis). In support for this inference, in vitro studies demonstrated that a number of genes regulating all that processes are modulated by IgG-aPL.
In conclusion, our overall data suggest that gene expression profiling allows the segregation of APS, APS plus SLE and SLE, with specific signatures explaining the pro-atherosclerotic, pro-thrombotic and inflammatory changes in these highly related autoimmune diseases.
Many autoimmune disorders have common features of disease pathogenesis.47 Although monocytes have been demonstrated to play a central role in atherosclerosis and CVD development, they are only one of the many players in the evolvement of these processes, which would further change depending on the presence of other traditional factors (age, obesity, diabetes, hypertension, etc.) and the probable influence of the specific therapy in each group of patients, such as glucocorticoids, antimalarials, antiplatelets, etc. Thus, a complex interplay of different environmental, therapeutic and genetic factors can determine different clinical expression in those diseases. Nevertheless, the identification of relevant genes whose products regulate specific physiological pathways might contribute to the development of targeted therapies for each autoimmune condition.
Acknowledgments
We thank all patients and healthy subjects for their participation in the study. We thank Ms Rosario Carretero for her excellent technical support.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
Handling editor Tore K Kvien
CP-S and NB shared first authorship.
MAA and CL-P shared last authorship.
Correction notice This article has been corrected since it was published Online First. The Funding section has been corrected.
Contributors SM, PR-L and CP-S developed the in vivo assays, performed the experiments and solved technical problems. MAA, MJC and MAK followed up with patients and contributed useful discussion and suggestions. AR-A, NB and CL-P formed the hypothesis, directed and coordinated the project, designed the experiments, analysed the data and wrote the manuscript. EC-E performed clinical analysis and contributed useful suggestions. YJ-G performed statistical analysis and discussed results.
Funding This work was supported by grants from the Junta de Andalucía (P08-CVI-04234 and CTS-7940), the Ministry of Health (PS09/01809 and PI12/01511), the Ministry of Science and Innovation (BFU2011-23578) of Spain, and the Spanish Rheumatology Foundation (FER). CL-P was supported by a contract from the Spanish Junta de Andalucía.
Competing interests None.
Patient consent Obtained.
Ethics approval This study was conducted with the approval of the Ethics Committee from the Reina Sofia Hospital from Cordoba-Spain.
Provenance and peer review Not commissioned; externally peer reviewed.