Article Text

Abstract
Objective Tofacitinib is an oral Janus kinase (JAK) inhibitor for the treatment of rheumatoid arthritis (RA). The pathways affected by tofacitinib and the effects on gene expression in situ are unknown. Therefore, tofacitinib effects on synovial pathobiology were investigated.
Methods A randomised, double-blind, phase II serial synovial biopsy study (A3921073; NCT00976599) in patients with RA with an inadequate methotrexate response. Patients on background methotrexate received tofacitinib 10 mg twice daily or placebo for 28 days. Synovial biopsies were performed on Days -7 and 28 and analysed by immunoassay or quantitative PCR. Clinical response was determined by disease activity score and European League Against Rheumatism (EULAR) response on Day 28 in A3921073, and at Month 3 in a long-term extension study (A3921024; NCT00413699).
Results Tofacitinib exposure led to EULAR moderate to good responses (11/14 patients), while placebo was ineffective (1/14 patients) on Day 28. Tofacitinib treatment significantly reduced synovial mRNA expression of matrix metalloproteinase (MMP)-1 and MMP-3 (p<0.05) and chemokines CCL2, CXCL10 and CXCL13 (p<0.05). No overall changes were observed in synovial inflammation score or the presence of T cells, B cells or macrophages. Changes in synovial phosphorylation of signal transducer and activator of transcription 1 (STAT1) and STAT3 strongly correlated with 4-month clinical responses (p<0.002). Tofacitinib significantly decreased plasma CXCL10 (p<0.005) at Day 28 compared with placebo.
Conclusions Tofacitinib reduces metalloproteinase and interferon-regulated gene expression in rheumatoid synovium, and clinical improvement correlates with reductions in STAT1 and STAT3 phosphorylation. JAK1-mediated interferon and interleukin-6 signalling likely play a key role in the synovial response.
Trial registration number NCT00976599.
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Cytokines play a critical role in the pathogenesis of rheumatoid arthritis (RA).1–3 Targeted biologics that block individual cytokines, such as tumour necrosis factor (TNF) or interleukin (IL)-6, have demonstrated clinical efficacy.4 Targeting intracellular pathways represents a novel approach of inhibiting the effects of multiple cytokines.5 ,6
Signal transduction allows the cell to sense cytokines in the external environment and initiate a cellular response.5 ,7 ,8 One example is the Janus kinase (JAK) family, which integrates signals from many cytokines.9 ,10 Four JAK proteins (JAK1, JAK2, JAK3 and tyrosine kinase 2 [Tyk2]) associate with the intracellular domains of surface cytokine receptors.9 ,10 Combinations of the JAK proteins allow site and event specificity, including JAK1/JAK3 for many T cell-derived cytokines, JAK1/JAK2 for IL-6, and JAK1/Tyk2 for interferons (IFNs).10 ,11 JAKs phosphorylate the signal transducers and activators of transcription (STATs) to modulate gene expression.10 ,12
The JAK inhibitor tofacitinib has proven effective in the treatment of RA.13–18 Tofacitinib is a targeted, small molecule inhibitor of several JAK isoforms, especially JAK3 and JAK1.19 We hypothesised that tofacitinib targets cytokine signalling critical to the pathogenesis of rheumatoid synovitis. Therefore, a double-blind, placebo-controlled serial synovial biopsy study was performed in patients with RA with an inadequate response to methotrexate. Treatment with tofacitinib reduced expression of matrix metalloproteinase (MMP) and IFN-regulated genes in rheumatoid synovium. Reduction of pSTAT1 and pSTAT3 measured in synovial biopsies at Month 1 was highly correlated with clinical improvement at Month 4. These data are consistent with tofacitinib acting on synovial JAK/STAT targets and suggest that JAK1-mediated signalling of IFNs and IL-6 plays a role in the synovial response to JAK blockade.
Methods
Study design and patients
A randomised, double-blind, placebo-controlled phase II, 28-day clinical study was performed across six centres in the USA (A3921073; NCT00976599). Patients were randomised 1:1 to tofacitinib 10 mg twice daily or placebo, all in combination with methotrexate. Patients were aged ≥18 years with active RA based on the American College of Rheumatology (ACR) 1987 Revised Criteria.20 Active disease was defined as ≥4 tender/painful and ≥4 swollen joints (of 68/66 joints examined), and either an erythrocyte sedimentation rate (ESR) >28 mm/h (Westergren method) or a C reactive protein level >7 mg/L. Patients had to have at least one knee, one elbow, one wrist or two metacarpophalangeal joints with active synovitis suitable for biopsy. Patients were receiving stable doses of 7.5–25 mg of methotrexate weekly and had an incomplete response.
Key exclusion criteria included: current treatment with other disease-modifying antirheumatic agents, including biologics; arthroscopy or intra-articular steroids within the previous 3 months; haemoglobin <9.0 mg/dL; absolute neutrophil count <1.2×109/L; recent, current or chronic infection; evidence of active, latent or inadequately treated Mycobacterium tuberculosis infection; or history of lymphoproliferative disorder or malignancy.
Patients were randomised in a 1:1 design to receive tofacitinib 10 mg twice daily or matching placebo tablets with background methotrexate. Approximately 4–10 days prior to the initiation of study drug, and at about Day 28 of dosing, the patients had arthroscopy under local anaesthesia or conscious sedation. The arthroscopy was performed on a clinically inflamed knee/elbow/wrist/metacarpophalangeal joint, selected by the investigator.
Patients from this study were eligible to enter an ongoing open-label, long-term extension (LTE) study (A3921024; NCT00413699; ongoing, 10 April 2013 data cut) 1 week after the second biopsy. Patients were followed up for both efficacy and safety within this LTE study and the results correlated with the Month 1 biopsy data for tofacitinib-treated patients.
This study was performed in compliance with the Declaration of Helsinki and Good Clinical Practice Guidelines established by the International Conference on Harmonisation. The final protocol, amendments and informed consent documentation were reviewed and approved by the institutional review board and the independent ethics committee of each investigational centre. All patients provided written informed consent.
Endpoints
The primary objective was to evaluate the effect of tofacitinib 10 mg twice daily on synovial tissue and blood biomarkers in active RA compared with placebo at Day 28. Secondary objectives were to evaluate the clinical efficacy, safety and tolerability of tofacitinib. The primary clinical outcomes were the disease activity score (DAS) based on a 28-joint count (DAS28-4 [ESR]) and the percentage of patients at Day 28 who met the criteria for an ACR20, ACR50 or ACR70 response (≥20%, ≥50% and ≥70% reduction of the composite ACR measures).2 Safety endpoints included the incidence and severity of adverse events and clinical laboratory abnormalities. DAS28 was also evaluated after patients had participated in the LTE study for 3 months. The DAS28 clinical efficacy evaluation was used to assess correlations with the biomarker changes observed on Day 28 of the A3921073 study. Additional clinical assessment was performed at 4 months from A3921073 baseline in the A3921024 study.
Synovial biopsies
Synovial tissue was acquired arthroscopically using automated motorised shaver technology. Pools of >10 tissue fragments each were aliquoted for histology, mRNA or protein analysis. For synovial immunohistochemistry, Optimal Cutting Temperature medium (OCT®) frozen blocks were made. Tissue fragments were frozen in an RNA STAT-60 for mRNA measurements. STAT protein analysis was performed on snap frozen tissue fragments.
Synovial gene expression analysis
Synovial RNA was extracted from pooled tissue fragments and cDNA was synthesised.21 Quantitative PCR (qPCR) was performed using predeveloped reagents (Applied Biosystems, Foster City, California, USA). Results were compared with a reference standard curve composed and normalised for glyceraldehyde 3-phosphate dehydrogenase.22 Post-treatment levels were expressed as the percentage of the pretreatment geometric mean with 95% confidence intervals from log-transformed data. The primary mRNA analysis included a limited number paired analytes evaluating inflammatory mediators, that is, cytokines (IL-1ß, TNF, IL-6), MMPs (MMP-1 and -3), IFN-regulated gene (ISG15) and chemokines (CCL2, CXCL10, CXCL13).
Synovial and serum ELISAs
Serum analytes were evaluated by enzyme-linked immunosorbent assay (ELISA) according to the manufacturer's instructions. Synovial tissue extracts for phosphorylated STAT were assayed by ELISA.
Synovial immunohistochemistry
DAKO (Glostrup, Denmark) reagents were used for immunohistochemistry according to the manufacturer's instructions on frozen sections (5 µm) fixed in acetone. Staining was quantified by digital image analysis using a Nikon E800 microscope and Image Pro software (AG Heinze, Lake Forest, California, USA). Data were reported as average percentage area stained in six fields.23 ,24
Statistical analysis
The clinical outcomes, both efficacy and safety, were summarised descriptively based on the intent-to-treat (ITT) population (patients who received ≥1 dose of randomised investigational drug). No imputation was performed for missing data. Synovial tissue and blood biomarkers were also summarised descriptively based on the ITT population.
The change from baseline (Day -7) at Day 28 in each parameter reported from the synovial tissue biopsy was compared between tofacitinib and placebo using an analysis of covariance model with treatment as a factor and baseline as a covariate. The change from baseline (Day 1 predose) at multiple postbaseline time points of each blood biomarker was compared between tofacitinib and placebo using a mixed-effects model with treatment, visit and treatment-by-visit interaction as fixed-effect factors, and baseline as a covariate. Correlation among repeated measurements at different visits on the same patient was modelled by including a common random effect in the aforementioned model. The multiple measurements on Day 1 were analysed separately from the multiple predose measurements taken on Days 10, 28, 29 and 35. No adjustments were made for multiple comparisons.
In addition, post hoc analyses were performed to: compute the percentage change, and associated t test-based p value, for within-treatment-group change from baseline; compute the correlation coefficient between the change in each biomarker from baseline (Day -7) to Day 28; and the change in DAS28 from Day 1 in the 28-day clinical study to Month 3 in the LTE study and its associated p value based on Fisher's transformation.
Results
Patients
A total of 15 patients were randomised to tofacitinib 10 mg twice daily plus methotrexate and 14 patients were randomised to placebo plus methotrexate. All randomised patients (N=29) were treated, completed the study and were analysed for efficacy and safety. There were no discontinuations from this study. All patients enrolled in the LTE study.
Patient demographic and baseline characteristics are shown in online supplementary table S1. The majority of patients were women (26/29) and Caucasian (23/29). The mean age of all patients was 53.3 years (range 27–77 years). The mean disease duration was 12.2 and 5.5 years in the tofacitinib and placebo groups, respectively. Patients in the tofacitinib group had greater disease severity in each of the ACR criteria components (mean scores), although the DAS28 was only slightly higher in the tofacitinib group.
Clinical responses at Day 28 and Month 4
Tofacitinib 10 mg twice daily led to decreases in DAS28 score compared with placebo on Day 28. Mean DAS28 scores decreased from baseline (6.55) to Day 28 (4.90) for patients receiving tofacitinib 10 mg twice daily compared with smaller decreases (6.03–6.32) in the placebo group (see table 1A for summary). After a total of 4 months' treatment (28 days in study 1073 and 3 months in LTE study), the tofacitinib group had a mean DAS28 of 4.77 and the placebo group 4.72 (see table 1B for summary of ACR and European League Against Rheumatism responses). DAS28 on Day 28 and 4 months were used for subsequent correlations between synovial analytes and clinical response.⇓
Clinical efficacy, as measured by EULAR response rate and ACR response rate, at (A) Day 28 and (B) after a total of 4 months' treatment
Safety
The safety experience was similar to previous tofacitinib studies. No serious adverse events were observed (see online supplementary table S2 for a summary of results). Three patients in the tofacitinib group and two patients in the placebo group had mild to moderate anaemia, and one patient receiving placebo had potentially life-threatening anaemia. No patient had neutrophil values of clinical concern.25
Effect of tofacitinib on synovial inflammation after 28 days
Changes in synovial cellular composition were measured by immunohistochemistry on baseline and Day 28 biopsies. No significant changes in CD3+ T cells, CD20+ B cells or CD68+ sublining macrophages were detected in tofacitinib or placebo groups (table 2 and figure 1).
IHC of synovial biopsy tissue

Absence of significant changes in CD68+ sublining macrophages. Representative synovial tissue samples were analysed by immunohistochemistry for CD68+ sublining macrophages at Day -7 (left) and Day 28 after tofacitinib treatment (right) (original magnification ×200).
Effect of tofacitinib on synovial gene expression after 28 days
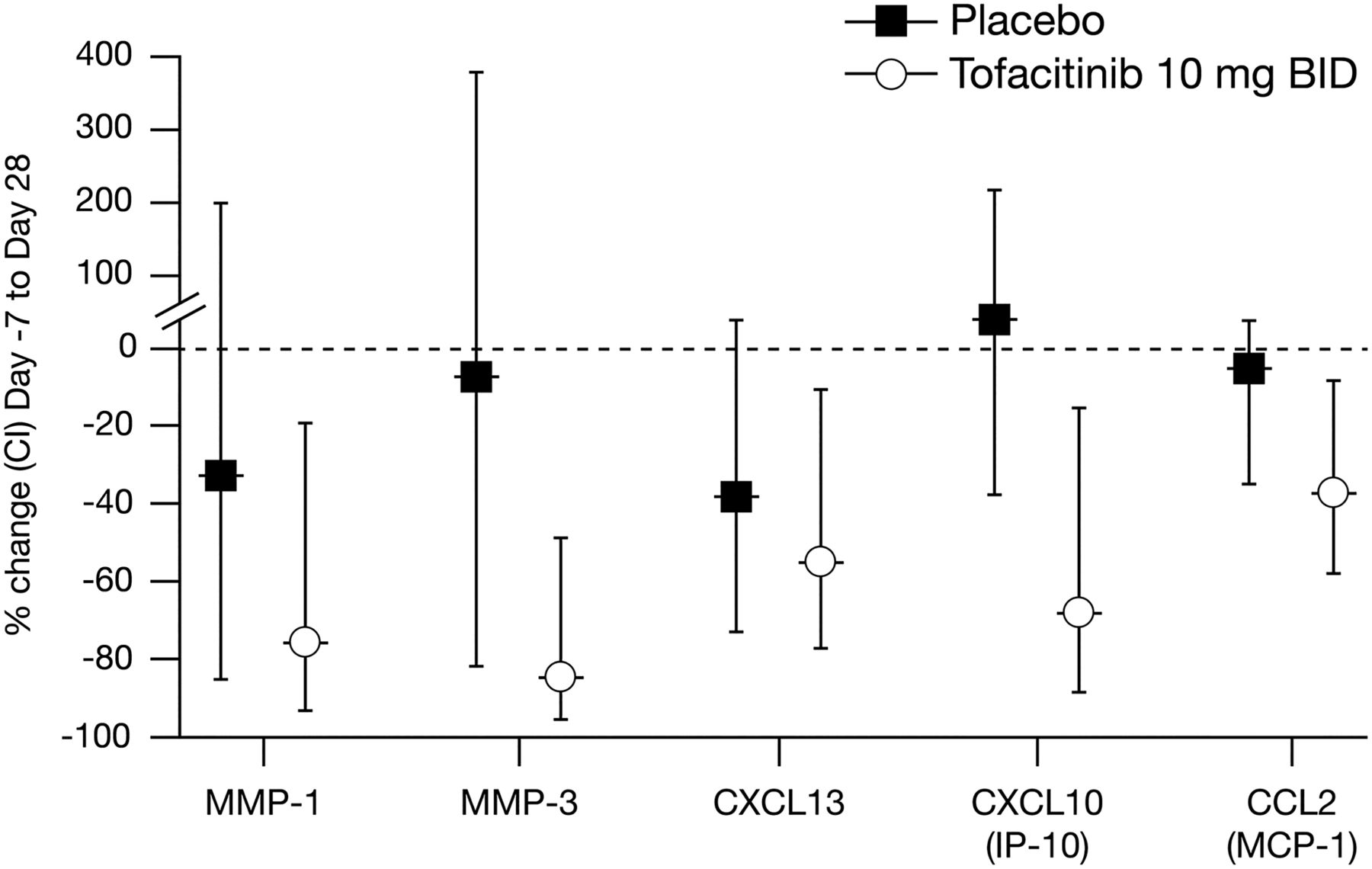
qPCR was performed on biopsies taken at baseline and Day 28 and changes in gene expression were evaluated. Key genes implicated in the pathogenesis of RA were significantly decreased in the tofacitinib group, including MMP-1, MMP-3, chemokine (C-X-C motif) ligand (CXCL)13, CXCL10 and chemokine (C-C motif) ligand (CCL)2 (figure 2). IL-6, ISG15 and TNF mRNA were not significantly changed on Day 28. No significant changes in gene expression were observed in the placebo-treated group.

Tofacitinib significantly reduced synovial chemokine and matrix metalloproteinase expression. BID, twice daily; CI, confidence interval; CCL, chemokine (C-C motif) ligand; CXCL, chemokine (C-X-C motif) ligand; IL, interleukin; IP-10, interferon gamma-induced protein 10; MCP-1, monocyte chemotactic protein-1; MMP, matrix metalloproteinase.
Change in synovial pSTAT levels and clinical response to tofacitinib
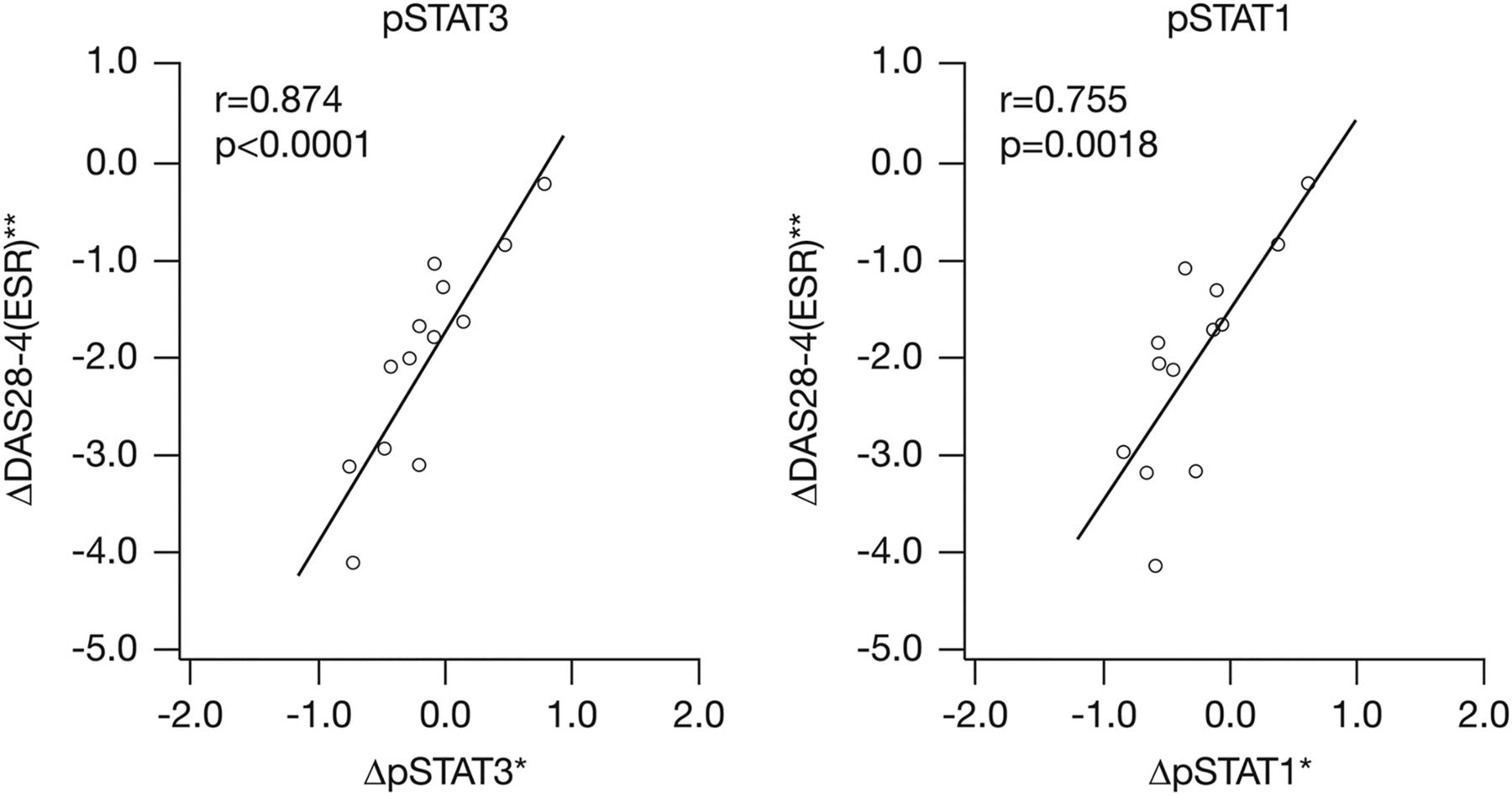
Clinical outcome data from the open-label LTE study on Month 3 was applied only to the tofacitinib group (13 patients received 10 mg twice daily and one patient received 5 mg twice daily in the LTE study), since the placebo group also received tofacitinib after LTE enrolment. The strongest correlation was an association between decreased synovial pSTAT levels after 1 month of tofacitinib treatment in the placebo-controlled study and clinical improvement after a total of 4 months including the LTE. STAT1 and STAT3 activation, as determined by phosphorylation of the transcription factors, were highly correlated with the change in DAS28 (r=0.755, p=0.0018 and r=0.874, p<0.0001, respectively) (figure 3). No changes in the levels of other biomarkers, including MMP-1, MMP-3, CXCL10 or CXCL2 correlated with clinical improvement.

Tofacitinib-induced synovial change in pSTAT3 and pSTAT1 at Day 28 predicts clinical response at 4 months. *Biomarker data change from baseline (Day -7) to Day 28. **DAS28-4(ESR) change from Day 1 to Month 4. DAS, disease activity score; ESR, erythrocyte sedimentation rate; pSTAT, phosphorylated signal transducer and activator of transcription; r, correlation coefficient; STAT, signal transducer and activator of transcription.
Relationship between baseline synovial biomarkers and clinical response
We then evaluated whether baseline mRNA biomarker expression and STAT phosphorylation predict clinical response (DAS28) after 4 months' treatment with tofacitinib. Only MMP-3 was statistically significant, with a higher pretreatment value correlating with improved response (p=0.0360; r=0.580).
Serum CXCL10 as a pharmacodynamic biomarker of tofacitinib activity
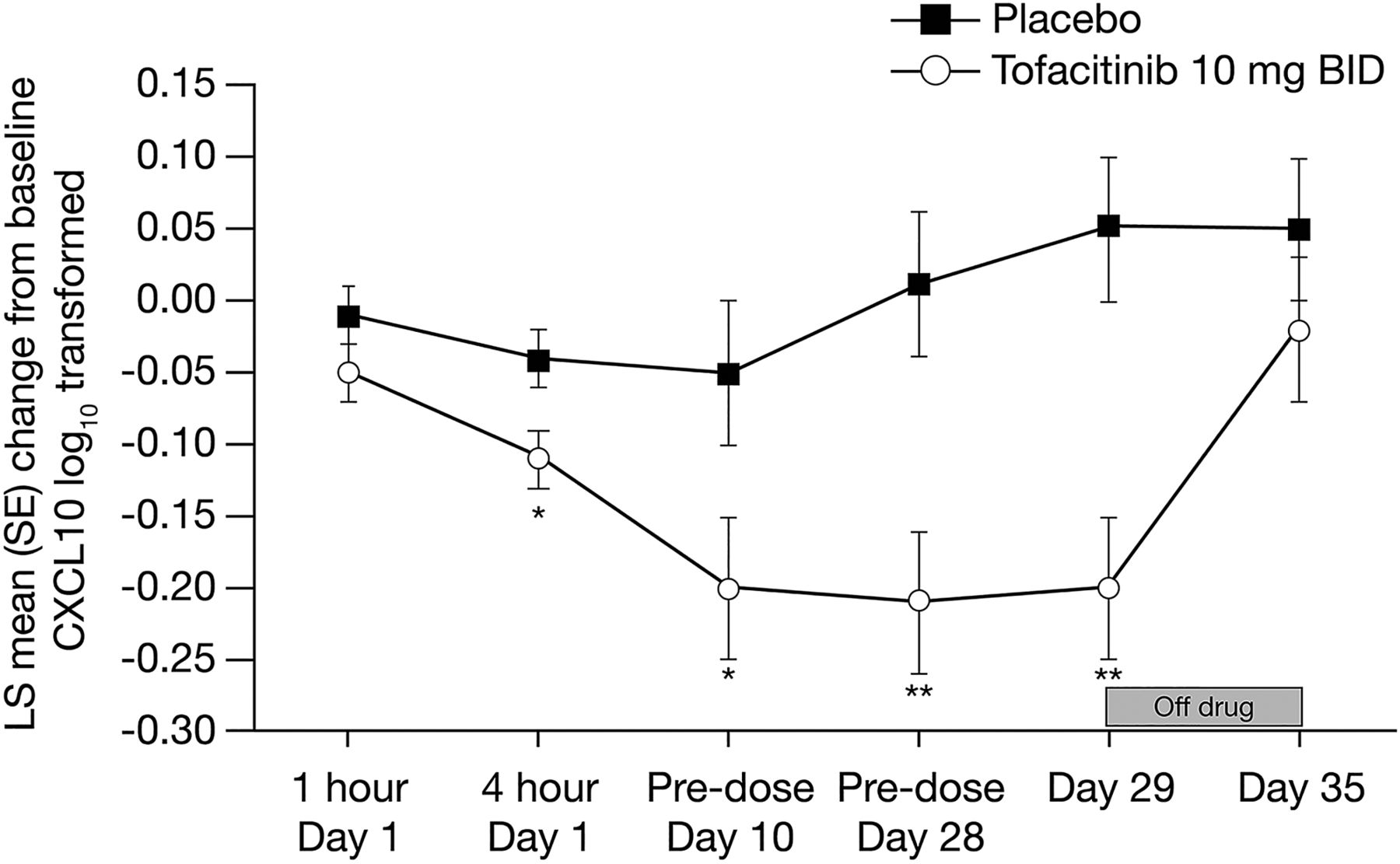
Serum chemokines were also evaluated at various times during the 28-day study. The most prominent finding was a rapid decrease in the IFN-regulated gene CXCL10, which was observed as early as 4 h after the first tofacitinib dose and which remained low throughout the treatment period (figure 4). One week after discontinuation of tofacitinib, CXCL10 levels returned to baseline. Both serum CXCL10 and synovial mRNA expression were reduced overall by tofacitinib activity; however, the levels at baseline or changes induced by tofacitinib did not correlate between serum and synovium (p>0.05). Furthermore, neither measure correlated with clinical response at 4 months (p>0.05).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
CXCL10 plasma levels in tofacitinib or placebo-treated patients with rheumatoid arthritis. *p<0.05, **p<0.005 change from Day 1 pre-dose relative to placebo. BID, twice daily; CXCL10, chemokine (C-X-C motif) ligand 10; LS, least squares; RA, rheumatoid arthritis; SE, standard error; LS means were calculated using two models; one for the measurements on Day 1, and one for the remaining measurements.
Discussion
We tested the hypothesis that tofacitinib targets cytokine signalling implicated in RA within the synovium. Tofacitinib reduced the synovial expression of MMPs and IFN-regulated genes in rheumatoid synovium. Analyses of the open-label LTE study 3-month efficacy data demonstrated a clear correlation between inhibition of synovial STAT1 and STAT3 activation after 1 month of therapy during the synovial biopsy portion of the study and the magnitude of clinical improvement assessed in the LTE (total 4 months of treatment with tofacitinib). These data suggest that JAK1-mediated signalling of IFNs and IL-6 plays a key role in the synovial response to JAK blockade.
Since cytokine inhibitors decrease disease activity in RA, blocking kinases in the signal transduction cascade could potentially have the same benefit. Targeting JAK proteins has met with success in clinical trials, and several JAK inhibitors have demonstrated efficacy in RA, similar to biological agents, in particular, tofacitinib.11 In addition to tofacitinib, these include several other JAK inhibitors with varying specificities for JAK isoforms, including baricitinib, VX509 and GLPG0634.11
The four JAK proteins (JAK1, 2, 3 and Tyk2) form a family of signalling molecules that associate with cytokine receptors.9 ,10 Combinations of JAK1 and/or JAK2 can transduce signals when members of the IL-6 family ligate their receptors, thereby activating their downstream targets, for example, STAT1 and STAT3.9 ,10 JAK1/JAK3 combinations facilitate signalling for cytokines that use the common gamma-chain receptor, particularly T cell cytokines.9 ,10 IFNs use several JAK/STAT combinations, but JAK1/Tyk2 and STAT1 are particularly important.9 ,10 Although the TNF receptor does not directly signal through JAKs, synoviocyte activation by TNF stimulation can be affected by JAK-dependent pathways.26
The relative contribution of specific JAK isoforms remains uncertain. In the present study, synovial biopsies were performed pretreatment and after 4 weeks of study treatment and correlated with clinical outcome. The selection of week 4 for the second biopsy and 4 months total exposure for clinical evaluation was based on our goal of identifying leading synovial biomarkers of clinical response rather than concomitant response. The 4-month time point allows comparison with the full clinical response.26
Unexpected issues in the current study were differences in disease duration and baseline severity between the two treatment groups, which were due to chance. The imbalance highlights the relatively small size of the study and the resulting limitations in statistical power which restricts the ability to perform stratification of subjects based on synovial architecture or clinical characteristics at baseline. This imbalance could contribute to differences when comparing synovial analytes in the placebo group with the tofacitinib group. Thus, relative changes of biomarkers within the groups were considered especially important.
Some therapeutic agents, such as TNF inhibitors and intra-articular steroids, cause a rapid and dramatic decrease in synovial inflammation;27 others show modest effects (methotrexate). Abatacept and rituximab cause minimal or no effect on the total cellular inflammation score at early time points.21 ,28 ,29 This is also the case with tofacitinib because cell infiltration is not significantly changed after 4 weeks of therapy. Individual cell lineages can be depleted with other agents, most notably B cells with rituximab; no significant effects of JAK inhibition on macrophage, B cell or T cell infiltration were observed at month 1.
The synovial biomarker study was notable for tofacitinib-induced decreases in expression of genes implicated in RA, including MMPs and chemokines. No significant effects were observed for any biomarker in the placebo group. Pretreatment levels of synovial biomarkers did not predict which patients would respond to tofacitinib and response was not associated with high or low relative synovial STAT phosphorylation at baseline.
Blood biomarkers were also assessed. A rapid and sustained decrease in CXCL10, which was also decreased in synovial tissue, was observed. Although CXCL10 serum protein and synovial mRNA levels were reduced by tofacitinib, changes in expression did not correlate to each other. CXCL10 is especially interesting because an anti-CXCL10 antibody has demonstrated some clinical benefit in RA.30 Therefore, the effect on this particular chemokine could be useful as a pharmacodynamic marker of tofacitinib activity. The role of CXCL10 as a type I IFN-induced and STAT-regulated chemokine suggests a role for IFN-mediated mechanisms. The ultimate utility of serum CXCL10 measurements could best be resolved in larger clinical studies.
Perhaps the most important finding of our study was the correlation between suppression of STAT activation, evidenced by STAT phosphorylation, and later clinical improvement. The pretreatment levels of pSTAT did not predict response to therapy possibly because the tofacitinib-treated group had greater disease severity. However, the declines in synovial pSTAT1 and pSTAT3 after 1 month were highly correlated with the degree of clinical response, as determined by change in DAS28. The correlation of change in pSTAT with clinical response, taken with the lack of change in synovial cellularity, suggests that the effect is transcription dependent. Further evaluations of tofacitinib inadequate responders, particularly those with high pSTATs at baseline, could provide insights into other roles of JAK-STAT signalling in RA.31
The biomarker data provide some clues to the mechanism of action of tofacitinib. Interfering with the JAK1-STAT3 pathway suggests that blocking IL-6 signalling contributes to the mechanism of action. However, the effects on pSTAT1 and IFN-regulated genes, such as CXCL10 production, suggest that the mechanism also involves blockade of IFN signal transduction. The potential importance of IFN blockade in clinical efficacy is supported by our recent observation that TNF-mediated synoviocyte activation is mediated by IFN production and JAK/STAT-induced expression of chemokines such as CXCL10.26 Therefore, tofacitinib can block IL-6 signalling directly and TNF function through an indirect mechanism. The contribution of individual synovial cell types to tofacitinib therapeutic sensitivity is still unknown and requires a larger study to evaluate the individual roles of B cells, T cells, macrophages and synoviocytes.
In conclusion, the data suggest that tofacitinib regulates JAK-mediated events in the rheumatoid synovium. Rather than blocking a single cytokine, tofacitinib modulates IL-6 and IFN signalling. JAK1 inhibition, with early suppression of STAT1 and STAT3, correlates well with longer-term clinical benefit. Finally, the effects of tofacitinib on blood chemokines such as CXCL10 suggest that biomarkers might be used to either predict or evaluate therapeutic response.
Acknowledgments
The authors would like to thank the patients who were involved in this study. Editorial support was provided by Anne Marie Reid, PhD, from Complete Medical Communications and funded by Pfizer Inc.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
Handling editor Tore K Kvien
Contributors Conception, design and interpretation: DLB, KS, JH, IK, SK, ZL, JB and GSF. Data collection: AK, DM, RS, NW, AKS, SR, DLB and PM. Data/statistical analysis: SK and ZL. Manuscript drafting: all authors.
Funding This study was sponsored by Pfizer Inc.
Competing interests DLB and AK have received research support from Pfizer Inc. NW and AKS have nothing to disclose. PM has received research support from, is a consultant for and has participated in a Speakers' bureau for Pfizer Inc. RS has received research support from and has participated in a Speakers' bureau for Pfizer Inc. DM and GSF are consultants for Pfizer Inc. KS, JH, IK, SK, ZL and JB are shareholders and employees of Pfizer Inc.
Ethics approval UCSD Institutional Review Board.
Provenance and peer review Not commissioned; externally peer reviewed.