Article Text

Abstract
Objective To evaluate whether subjects with knee or hip osteoarthritis (OA) pain on non-steroidal anti-inflammatory drugs (NSAIDs) received greater benefit when tanezumab monotherapy replaced or was coadministered with NSAIDs.
Methods Subjects (N=2700) received intravenous tanezumab (5 or 10 mg) or placebo every 8 weeks with or without oral naproxen 500 mg twice daily or celecoxib 100 mg twice daily. Efficacy was assessed as change from baseline to week 16 in three co-primary endpoints: Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) Pain, WOMAC Physical Function and Patient's Global Assessment (PGA) of OA. Safety assessments included adverse events, physical and neurological examinations, laboratory tests and vital signs.
Results Although all tanezumab treatments provided significant improvements in WOMAC Pain and Physical Function over either NSAID alone, only tanezumab+NSAIDs were significant versus NSAIDs with PGA and met the prespecified definition of superiority. Combination treatment did not substantially improve pain or function over tanezumab monotherapy. Adverse event frequency was higher with tanezumab than with NSAIDs and highest with combination therapy. Higher incidence of all-cause total joint replacements occurred with tanezumab+NSAID versus tanezumab monotherapy or NSAIDs. Rapidly progressive OA incidence was significantly greater versus NSAID in all tanezumab groups except tanezumab 5 mg monotherapy.
Conclusions Subjects receiving partial symptomatic relief of OA pain with NSAIDs may receive greater benefit with tanezumab monotherapy. While only coadministration of tanezumab with NSAIDs met the definition of superiority, combination treatment did not provide important benefits over tanezumab monotherapy; small differences in efficacy were negated by treatment-limiting or irreversible safety outcomes.
Trial registration number NCT00809354
Statistics from Altmetric.com
Introduction
Tanezumab is a humanised monoclonal antibody with high affinity and selectivity for the pain-mediating neurotrophin nerve growth factor (NGF).1 ,2 Efficacy of this compound in relieving chronic pain appears promising.3–10 In clinical trials of subjects with osteoarthritis (OA) pain, tanezumab consistently improved pain, function and patient global assessments of disease activity versus placebo or naproxen treatment.3–5 ,8 ,9 Recently, we reported that tanezumab added to a stable diclofenac sustained release regimen provided significantly greater efficacy than diclofenac plus placebo but was associated with higher adverse event incidence.11
To extend results of prior tanezumab studies, we examined efficacy and safety of tanezumab administered either alone (monotherapy) or in combination with a non-steroidal anti-inflammatory drug (NSAID; naproxen 500 mg twice daily or celecoxib 100 mg twice daily) versus NSAID alone for treatment of OA pain. The objectives of this study were to determine (1) whether subjects receiving only partial symptomatic relief of OA pain on a stable naproxen or celecoxib regimen received greater benefit after switching to tanezumab and (2) whether addition of tanezumab to a stable NSAID regimen provided additional benefit. This study differs from previous studies in the choice of active comparators and the study design that allows evaluation of tanezumab monotherapy and tanezumab+NSAIDs simultaneously.
Methods
This was a double-blind, parallel-group, multicentre, randomised clinical trial of subjects with OA pain of the knee or hip receiving only partial pain relief from a stable naproxen or celecoxib regimen but otherwise tolerating treatment. The study was conducted in compliance with the Declaration of Helsinki and all International Conference on Harmonisation Good Clinical Practice guidelines and registered at clinicaltrials.gov (NCT00809354). The protocol and informed consent documentation were reviewed and approved by Institutional Review Boards or Ethics Committees. Subjects provided written informed consent before protocol-specified procedure initiation.
Study population
Eligible subjects were 18 years of age or older, had a diagnosis of knee or hip OA based on American College of Rheumatology criteria with radiographic confirmation (Kellgren–Lawrence grade ≥2 on a scale of 0–4),12 ,13 body mass index ≤ 39 kg/m2, taking stable oral NSAID (naproxen (500–1000 mg/day) or celecoxib (200 mg/day)) for their OA pain for a minimum of 30 days prior to screening, and experiencing at least some analgesic benefit from the NSAID in the opinion of the investigator. Subjects were required to report Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) Pain score ≥4 (on an 11-point numeric rating scale (NRS)) at screening to qualify for the study. To be eligible for randomisation, subjects were required to report a WOMAC Pain score ≥4; WOMAC Physical Function score ≥4 (on an 11-point NRS); overall condition of fair, poor or very poor on the Patient's Global Assessment (PGA) of OA at baseline; and have at least 70% compliance with study-supplied oral NSAID treatment over at least 14 days directly prior to baseline. Since subjects were required to continue stable oral NSAID use prior to randomisation, subjects were not required to report a worsening WOMAC Pain score (ie, no ‘flare’).
Key exclusion criteria were similar to other tanezumab trials3 ,4 but also included any abnormality that would preclude continued NSAID therapy, including country-specific restrictive exclusion criteria for naproxen or celecoxib use in subjects with cardiac disease.
Study design
The study consisted of a screening period of up to 30 days (for discontinuation and washout of all prohibited pain medications with continuation of study-supplied NSAID), a 56-week treatment period and an 8-week follow-up period (see online supplementary figure S1). Study visits were conducted at screening, baseline (randomisation; day 1) and weeks 2, 4, 8, 12, 16, 24, 32, 40, 48, 56 and 64. General health and well-being, safety and concomitant medication use were assessed by telephone at weeks 20, 28, 36, 44 and 52.
Subjects were stratified by prestudy NSAID and index joint (hip or knee) and randomised in equal allocation in two cohorts (naproxen or celecoxib). The study was conducted as one protocol with these cohorts with a prespecified design to enrol a minimum of 500 subjects in each treatment (a total of 2500 subjects) with a minimum of 210 subjects per treatment within each cohort (see online supplementary text 1). Subjects were randomised according to a computer-generated randomisation code to one of five treatments: (1) intravenous tanezumab 5 mg and matching oral placebo for NSAID; (2) intravenous tanezumab 10 mg and oral placebo for NSAID; (3) intravenous tanezumab 5 mg and naproxen 500 mg twice daily or celecoxib 100 mg twice daily; (4) intravenous tanezumab 10 mg and naproxen 500 mg twice daily or celecoxib 100 mg twice daily; or (5) intravenous placebo for tanezumab and naproxen 500 mg twice daily or celecoxib 100 mg twice daily. Subjects randomised to tanezumab 5 mg or 10 mg monotherapy were tapered off prestudy NSAID in a blinded manner over 2 weeks following their first dose of intravenous study medication. Tanezumab or matching placebo was administered intravenously at baseline and then every 8 weeks to a maximum of seven administrations. In the event of inadequate pain relief, supplied rescue medication (500 mg acetaminophen; maximum of 4000 mg/day) could be used up to 3 days/week, but had to be discontinued at least 48 h prior to any study visit.
During this study, the US Food and Drug Administration (FDA) placed all clinical studies of tanezumab except those evaluating cancer pain on clinical hold due to unexpected adverse events initially described as osteonecrosis that required total joint replacement (TJR) (see online supplementary text 2). The primary efficacy objectives of this study were not impacted by the clinical hold, although assessment of long-term efficacy (beyond 16 weeks) was limited. With regard to safety objectives, not all subjects had the opportunity to receive treatment for 1 year as originally planned.
Efficacy
Three co-primary efficacy outcomes were predefined: change from baseline to week 16 for WOMAC Pain and Physical Function (recorded on an 11-point NRS; higher scores indicate greater pain or worse physical function)14 and PGA of OA (recorded on a five-point scale (1=very good to 5=very poor)).15 The landmark analysis was not changed due to the clinical hold.
Secondary efficacy measures included mean change from baseline to other assessment times for WOMAC Pain, percentages of subjects with ≥30%, ≥50%, ≥70% and ≥90% reductions from baseline on WOMAC Pain subscale at week 16, and percentages of subjects with an Outcome Measures in Rheumatology-Osteoarthritis Research Society International (OMERACT-OARSI) response at week 16.16 All efficacy outcomes were assessed at study visits except rescue medication use, which was reported daily via an interactive voice response system.
Safety
Safety assessments included onset, duration, severity and outcome of adverse events; physical and neurological examinations; clinical laboratory tests; and vital signs. After implementation of the FDA clinical hold, subjects were followed for protocol-specified safety assessments for at least 16 weeks after last dose of intravenous study medication. Study investigators performed standardised neurological examinations at each visit.17 ,18 If a neurological adverse event or clinically significant change on neurological examination occurred, the subject was referred to a neurologist for further evaluation.
Following the FDA clinical hold, an independent Adjudication Committee was formed to review all reported adverse events described by investigators as osteonecrosis and all reported TJRs including those unrelated to osteonecrosis.19 ,20
Statistical analysis
Efficacy and safety were assessed in all randomised subjects receiving at least one intravenous tanezumab or placebo dose (intent-to-treat population). Efficacy assessments were made separately within each NSAID cohort. Once comparable safety results were confirmed for each cohort separately, safety results were combined.
Baseline observation carried forward (BOCF) imputation was the primary analysis used for missing data. Co-primary efficacy endpoints were analysed using an analysis of covariance (ANCOVA) model, with model terms for baseline score and index joint. Least squares means were used for treatment effects and comparisons estimates. Fixed-sequence testing ordered by tanezumab dose was used for evaluation of all co-primary efficacy endpoints. The first comparison was tanezumab 10 mg+NSAID versus NSAID alone. If this analysis was significant (two-sided p<0.05), then tanezumab 5 mg+NSAID versus NSAID and tanezumab 10 mg versus NSAID were tested simultaneously using the Hochberg procedure. If both comparisons were statistically significant, then tanezumab 5 mg versus NSAID was tested. A treatment contrast was declared superior only if comparisons were significant for all three co-primary endpoints. The same ANCOVA model and BOCF imputation were used for analyses of change from baseline to weeks 2, 4, 8 and 12 on the WOMAC Pain subscale.
Results
From February 2009 through December 2009, 4531 subjects were screened for eligibility and 2700 received at least one tanezumab or placebo treatment (figure 1). All subjects treated with one or more doses of intravenous study medication were evaluated for efficacy and safety with 79.9–88.3% of subjects receiving two doses of blinded intravenous study medication and completing week 16 efficacy assessments. Subject baseline and demographic characteristics were comparable across treatments and cohorts (table 1). Study termination due to the clinical hold caused 48.7% (1314/2700) of all subjects to discontinue. Discontinuations due to lack of efficacy were highest among subjects treated with NSAID monotherapy, whereas discontinuations due to adverse events were highest among subjects treated with tanezumab 10 mg+NSAID. Study medication exposure was similar across treatments and between cohorts (see online supplementary tables S1 and S2).
Subject demographics and baseline characteristics
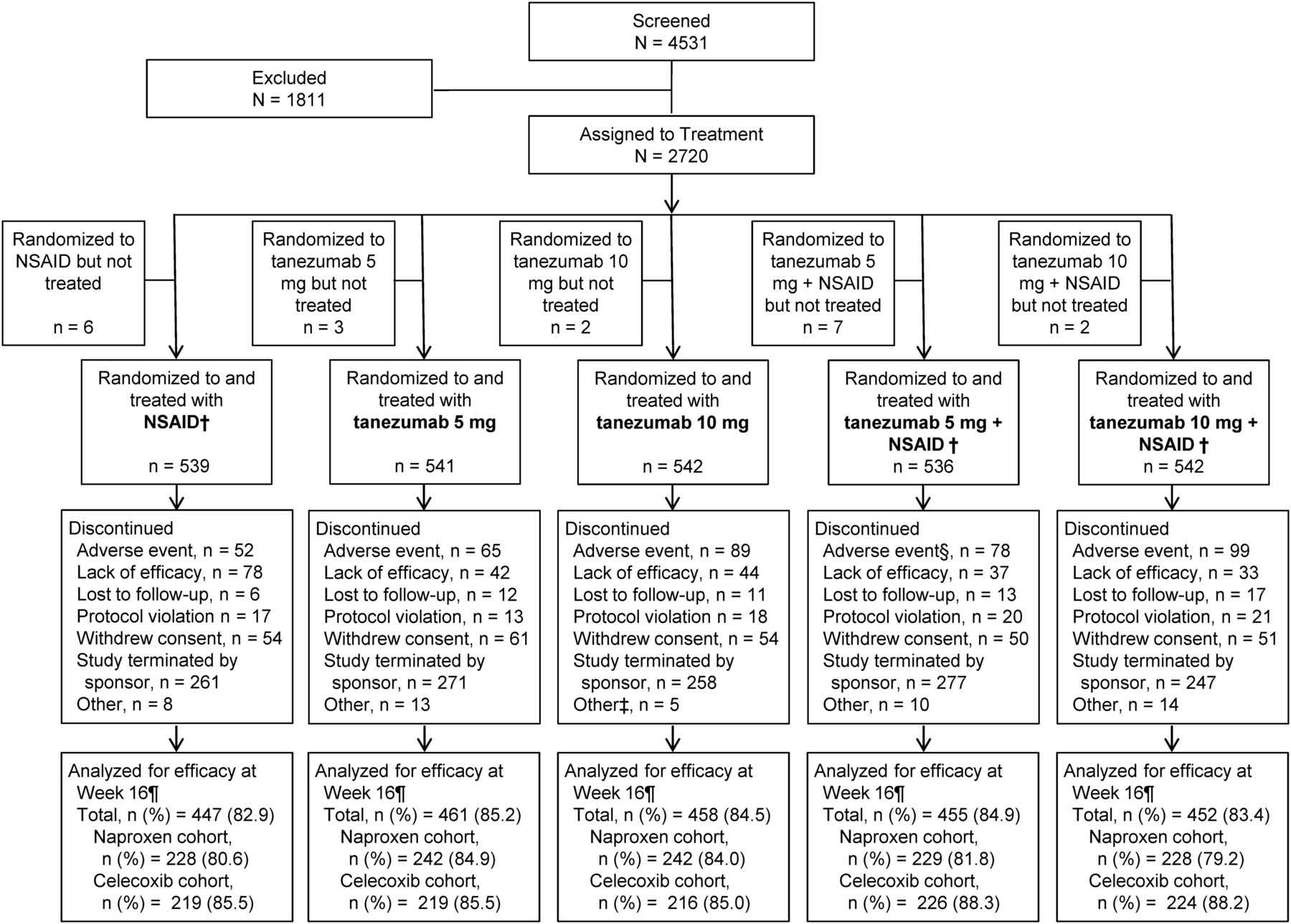
Subject disposition.* Excluding subjects who were withdrawn from the study due to termination of the study due to the clinical hold, the percentage of subjects that discontinued the study overall (both prior and after the clinical hold) ranged from 38.1% to 43.4% across the treatment groups. †Naproxen 500 mg twice daily or celecoxib 100 mg twice daily. ‡Other includes one subject who withdrew due to pregnancy. §Adverse event includes one subject in the celecoxib cohort who died. ¶Number of patients with observed data change from baseline at week 16. NSAID, non-steroidal anti-inflammatory drug.
Efficacy
Tanezumab monotherapy
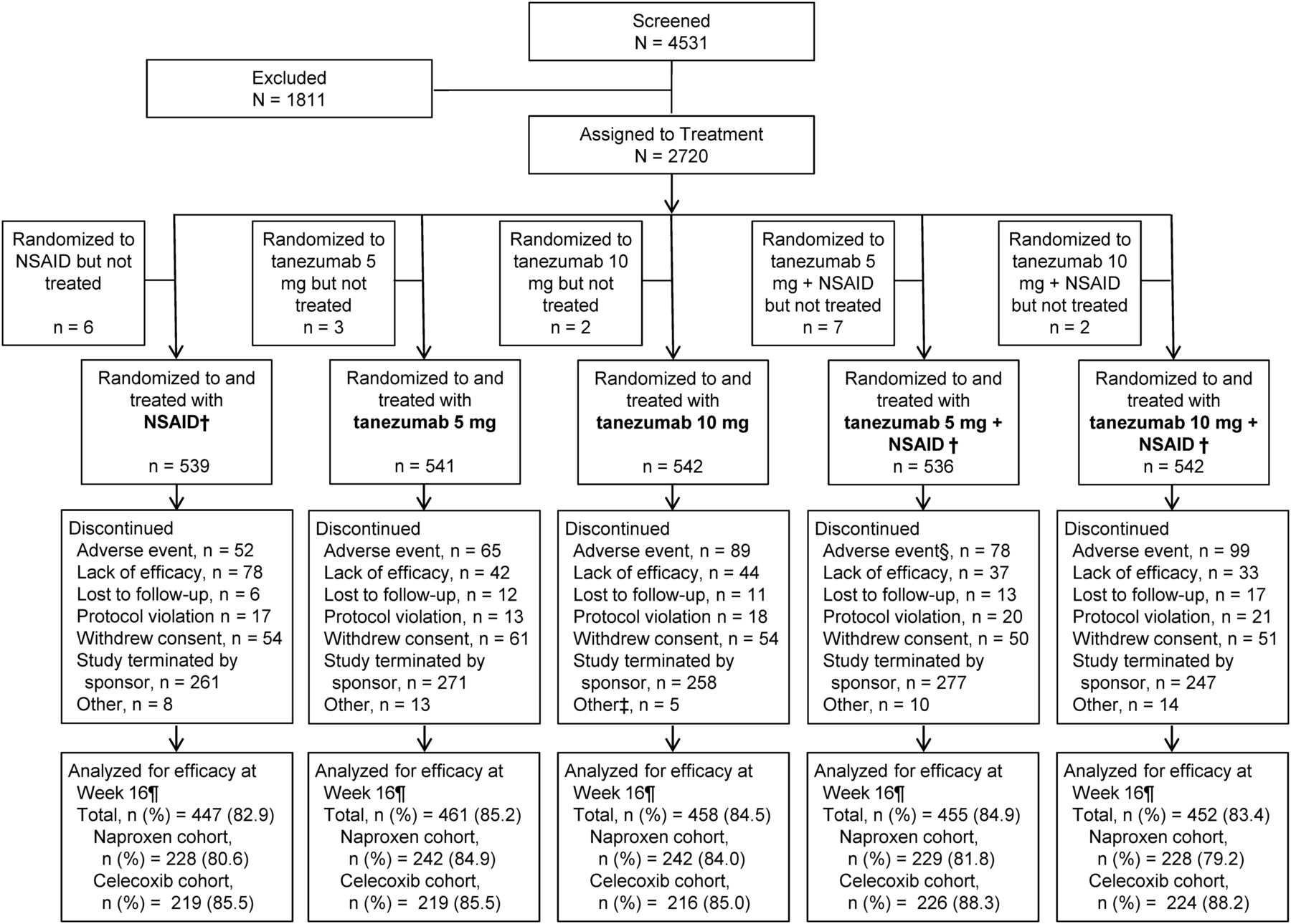
At the 16-week landmark timepoint, tanezumab 5 and 10 mg resulted in significantly greater mean improvement of WOMAC Pain and Physical Function compared with naproxen (p≤0.015; figure 2A; see online supplementary table S3) and celecoxib (p≤0.007; figure 2B; see online supplementary table S3). Both tanezumab monotherapy treatments provided consistent and sustained improvements in WOMAC Pain over either NSAID alone beginning 4 weeks after the initial dose of tanezumab (see online supplementary figure S2A,B). For PGA of OA, tanezumab monotherapy resulted in improvements versus NSAIDs alone (tanezumab 5 mg vs celecoxib and tanezumab 10 mg vs either NSAID), but treatment differences did not reach statistical significance (p>0.05; figure 2A,B; see online supplementary table S3), therefore neither dose met the definition of superiority versus either NSAID. Improvements in all co-primary endpoints were generally similar between tanezumab 5 mg and tanezumab 10 mg groups.

Change from baseline to week 16 in the WOMAC Pain subscale, WOMAC Physical Function subscale and Patient's Global Assessment of OA (using BOCF imputation for missing data). (A) Naproxen cohort. (B) Celecoxib cohort. *p≤0.05; **p≤0.01; ***p≤0.001 versus NSAID; +p≤0.05: tanezumab 10 mg+NSAID versus tanezumab 10 mg; naproxen dose=500 mg twice daily; celecoxib dose=100 mg twice daily. BOCF, baseline observation carried forward; NSAID, non-steroidal anti-inflammatory drug; OA, osteoarthritis; WOMAC, Western Ontario and McMasters Universities Osteoarthritis Index.
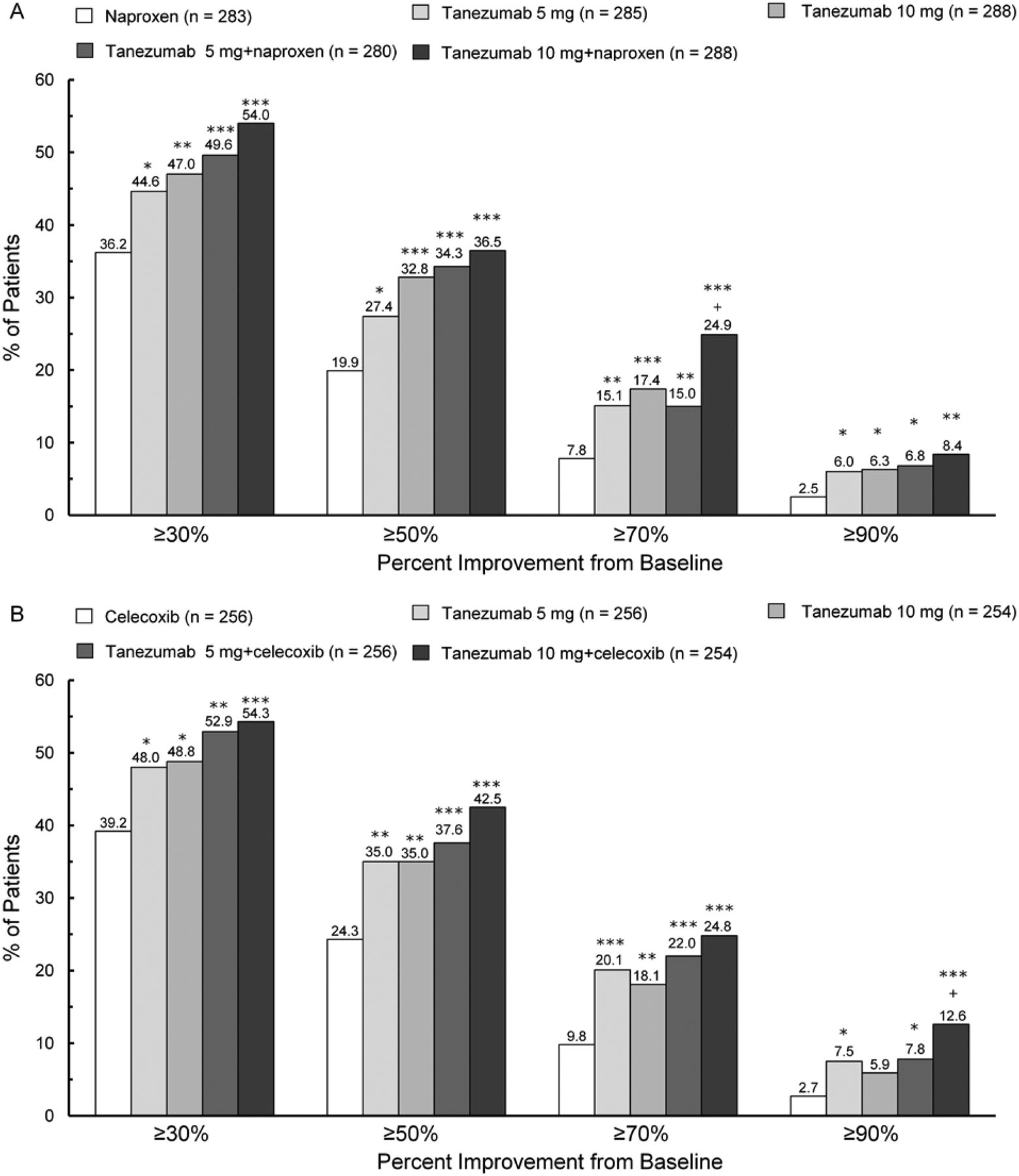
Compared with subjects treated with either NSAID alone, more subjects receiving tanezumab 5 or 10 mg monotherapy had clinically significant treatment responses at week 16. Percentages of subjects with ≥30%, ≥50%, ≥70% and ≥90% reductions in WOMAC Pain from baseline to week 16 were significantly greater with tanezumab monotherapy than with either NSAID alone (p≤0.044) for all comparisons except tanezumab 10 mg versus celecoxib at the 90% level (figure 3A,B). For OMERACT-OARSI outcomes, percentages of subjects with treatment response at week 16 were higher with tanezumab monotherapy (range 52.3–55.9%) than with either NSAID alone (naproxen 45.2%; celecoxib 47.3%), although only tanezumab 10 mg versus celecoxib met statistical significance (p=0.049; data not shown).

Percentages of subjects with a response ≥30%, ≥50%, ≥70% and ≥90% from baseline on WOMAC Pain subscale at week 16. (A) Naproxen cohort. (B) Celecoxib cohort. *p≤0.05; **p≤0.01; ***p≤0.001 versus NSAID; +p≤0.05: tanezumab 10 mg+NSAID versus tanezumab 10 mg. Naproxen dose=500 mg twice daily; celecoxib dose=100 mg twice daily. NSAID, non-steroidal anti-inflammatory drug; WOMAC, Western Ontario and McMasters Universities Osteoarthritis Index.
Tanezumab+NSAID combination therapy
Tanezumab+NSAID treatments resulted in significantly greater mean improvement versus either NSAID alone in all co-primary endpoints for all comparisons except tanezumab 5 mg+naproxen versus naproxen for PGA (figure 2A,B). As a result, tanezumab 10 mg in combination with naproxen or celecoxib and tanezumab 5 mg+celecoxib met the definition of superiority. Addition of tanezumab to naproxen or celecoxib rarely resulted in greater efficacy than the same doses administered as monotherapy. As noted with tanezumab monotherapy, all tanezumab+NSAID treatments provided consistent and sustained efficacy over NSAIDs alone beginning 4 weeks after the initial dose (see online supplementary figure S2A,B). In addition, at week 16, significantly larger percentages of subjects receiving tanezumab+NSAID had ≥30%, ≥50%, ≥70% and ≥90% reductions from baseline in WOMAC Pain compared with either NSAID alone (p≤0.015; figure 3A,B) and clinically meaningful OMERACT-OARSI treatment responses (range 56.8–63.4% vs 45.2% with naproxen and 47.3% with celecoxib; p≤0.005; data not shown).
Safety
The naproxen and celecoxib cohorts were combined for safety assessment. Overall, frequency of adverse events was greater among tanezumab-treated subjects than in subjects receiving NSAIDs (table 2). Adverse event frequency was similar between tanezumab 5 and 10 mg, and generally no differences were observed when tanezumab was administered as monotherapy versus in combination with NSAIDs. Peripheral oedema, paraesthesia and hypoesthesia were more common in subjects treated with tanezumab+NSAID than with tanezumab monotherapy, whereas the opposite pattern occurred for arthralgia and joint swelling. Paraesthesia and pain in extremity were the only adverse events that increased in frequency with tanezumab 10 vs 5 mg whether administered as monotherapy or combined with an NSAID.
Summary of safety including incidence of most frequent (≥3% in any treatment group) adverse events
Overall incidence of withdrawals due to adverse events was greater among tanezumab-treated subjects compared with subjects treated with NSAID alone and highest in subjects receiving tanezumab 10 mg (monotherapy or combined with NSAIDs; table 2). Incidence of serious adverse events was similar with tanezumab 5 mg, tanezumab 10 mg and NSAIDs; tanezumab combined with NSAIDs was associated with the highest frequency of serious adverse events. Worsening OA, reported osteonecrosis and arthralgia were the most commonly reported serious adverse events in all treatments. Incidence of these adverse events was similar across the tanezumab 5 mg, tanezumab 10 mg and NSAID groups. Tanezumab+NSAID treatment was associated with the highest frequency for worsening OA and reported osteonecrosis but not for arthralgia, where frequency was highest in the NSAID group.
Among the most frequently reported serious adverse events that were not joint-related and were reported in ≥2 subjects in at least one tanezumab group but not reported with NSAID alone were tibia fracture, femur fracture, atrial fibrillation, pulmonary embolism, gastric cancer, anaemia, upper gastrointestinal haemorrhage, dizziness, spinal fracture and hypertension. Among the most frequently reported serious adverse events, arthralgia, chest pain and cholelithiasis were reported more frequently with NSAID alone than in any tanezumab group.
Five subjects died during the study: two in the tanezumab 5 mg group due to pulmonary embolism and metastatic pancreatic cancer, respectively; one each in the tanezumab 5 mg+naproxen, tanezumab 5 mg+celecoxib and tanezumab 10 mg+celecoxib groups due to metastatic non-Hodgkin’s lymphoma, gastric cancer and lung cancer with cardiopulmonary arrest, respectively. None of the deaths was considered by investigators to be related to study medication.
Among adverse events associated with abnormal peripheral sensation, paraesthesia, hypoesthesia, burning sensation, peripheral neuropathy, decreased vibratory sense and hyperaesthesia were reported by ≥1% more subjects receiving any tanezumab treatment than in the NSAID group. More subjects had clinically significant changes in their final neurological examination with tanezumab (1.3–2.4%) than with NSAIDs (0.9%; see online supplementary table S4). Similarly, categorisations of final neurological consultations as suggestive of new or worsened peripheral neuropathy from diagnostic testing or clinically significant signs on neurologist examination were similar across tanezumab groups (5.7–6.6%) and more common than in subjects receiving NSAID alone (1.7%).
Source documents from 33 of 33 (100%) subjects with reported adverse events of osteonecrosis and 73 of 117 (62.4%) subjects with TJR unrelated to osteonecrosis identified in this study were obtained and reviewed by the Adjudication Committee. Consensus was reached on all cases adjudicated in this study. Only one event was adjudicated as primary osteonecrosis. Based on the available radiology, the Adjudication Committee estimated that the primary osteonecrosis had been present for more than 1 year prior to the reporting of this adverse event. Adjudication outcomes in the remaining subjects were worsening OA (n=92), other diagnosis (n=10) or insufficient information to distinguish osteonecrosis from worsening OA (n=3). Of the 92 subjects adjudicated with worsening OA, 56 were adjudicated with normal progression of OA, 34 were adjudicated with rapidly progressive OA and there was insufficient information for two subjects to allow the committee to make a determination between normal and rapid progression of OA.
Incidence of rapidly progressive OA was 0.7% with tanezumab 5 mg and 1.3% with tanezumab 10 mg versus 0.2% with NSAID alone. HRs (95% CI) determined by Cox proportional hazards model of time to an adjudicated event of rapidly progressive OA with tanezumab 5 or 10 mg monotherapy versus NSAID alone were 4.3 (0.5 to 38.4; p=0.197) and 7.3 (0.9 to 60.1; p=0.064), respectively. In tanezumab+NSAID groups, incidence of rapidly progressive OA was 1.7% with tanezumab 5 mg+NSAID and 2.4% with tanezumab 10 mg+NSAID with HRs versus NSAID alone of 10.4 (1.3 to 83.1; p=0.026) and 10.2 (1.3 to 80.0; p=0.028), respectively.
Of the 34 adjudicated events of rapidly progressive OA, 19 subjects were adjudicated as unilateral rapidly progressive OA of the hip (two of these had rapidly progressive OA of one hip and worsening OA in the contralateral hip), 13 subjects adjudicated as unilateral rapidly progressive OA of the knee (three of these were adjudicated as rapidly progressive OA in one knee and worsening OA in the contralateral knee) and 1 subject had unilateral rapidly progressive OA of the shoulder. One subject treated with NSAID monotherapy was adjudicated with bilateral knee rapidly progressive OA. Nineteen of the twenty-eight subjects with unilateral rapidly progressive OA experienced the event in the index joint while the remaining nine events occurred in the non-index joint. In the six subjects with bilateral events, three had rapidly progressive OA in the index joint and normally progressing OA in the contralateral joint, one had normally progressing OA in the index joint and rapidly progressive OA in the contralateral joint, one had rapidly progressive OA in the hip when the index joint was the left knee and one had rapidly progressive OA in both the index knee and contralateral knee. Of the rapidly progressive OA events adjudicated in the index joint, five were Kellgren–Lawrence grade 2, nine were Kellgren–Lawrence grade 3 and nine were Kellgren–Lawrence grade 4. Based on review of available prestudy radiology images, the Adjudication Committee identified four subjects with pre-existing rapidly progressive OA at the time of study entry (one each across the four tanezumab groups).
Rates of all-cause TJR were up to twofold greater with tanezumab+NSAID than with tanezumab monotherapy or NSAID (figure 4A); all-cause TJR occurred at similar rates in subjects treated with tanezumab monotherapy and NSAID alone. Percentages of subjects experiencing events of rapidly progressive OA (as determined by the Adjudication Committee) were elevated in all tanezumab groups relative to subjects receiving NSAID alone (figure 4B). The event rate increased as a function of the tanezumab dose, and administration of tanezumab combined with NSAID further increased the rate of rapidly progressive OA twofold over tanezumab monotherapy.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Percentages of subjects with events. (A) All-cause total joint replacements. (B) Rapidly progressive osteoarthritis (OA). *As determined by the adjudication committee.
Discussion
This randomised controlled study was designed to compare efficacy and long-term safety of tanezumab as monotherapy or in combination with an NSAID to NSAID treatment alone in subjects with hip or knee OA pain treated with stable naproxen or celecoxib prior to study entry. Despite NSAID treatment, subjects entered the study with baseline mean WOMAC Pain scores of approximately 6.4, which is just below the level of severe pain (≥7) on the 11-point NRS, indicating that NSAIDs alone were providing suboptimal pain relief. When re-randomised to NSAID monotherapy treatment (same NSAID) in a blinded fashion, subjects recorded modest pain relief from baseline (see online supplementary text 3). In comparison, administration of tanezumab 5 mg or tanezumab 10 mg monotherapy or in combination with naproxen or celecoxib provided greater mean pain improvement that was significant versus either NSAID alone. All tanezumab treatments also demonstrated significant improvements in WOMAC Physical Function compared with either NSAID alone. For patient global scores, improvements observed with tanezumab monotherapy did not reach statistical significance for either dose tested when compared with NSAID treatment; combination of tanezumab treatment with an NSAID resulted in significant improvements with both tanezumab doses in the celecoxib cohort and the tanezumab 10 mg dose in the naproxen cohort. Because of the definition for superiority employed, which required statistically significant improvements across all co-primary endpoints, only tanezumab+NSAID combination therapy (specifically, tanezumab 10 mg combined with naproxen or celecoxib and tanezumab 5 mg+celecoxib) was superior to treatment with an NSAID alone. Improvement with combination treatment was also consistently observed with the OMERACT-OARSI responder index (see online supplementary text 4). On average, 1 in 3 tanezumab treated-subjects had ≥50% reduction in pain and 1 in 15 subjects reported ≥90% pain reduction. In terms of group mean responses, maximal analgesic efficacy was observed following all tanezumab treatments 4 weeks following the initial administration. This level of response was maintained through the end of the first 8-week dosing interval and throughout the second 8-week dosing interval.
While significant improvement occurred with tanezumab combined with NSAIDs, the co-primary endpoints of pain and function did not differ significantly from what was achieved with tanezumab monotherapy. Further, there were more adverse events with tanezumab+NSAID than with tanezumab monotherapy or NSAID alone. Similarly, tanezumab 10 mg monotherapy did not provide substantial improvements in efficacy over tanezumab 5 mg monotherapy.
Overall adverse event incidence and withdrawals due to adverse events were generally similar among groups, although rates in tanezumab groups tended to be higher than with NSAIDs alone. Serious adverse event frequencies with tanezumab monotherapy were similar to NSAIDs alone. Differences in frequency were largely driven by four adverse events: arthralgia, paraesthesia, hypoesthesia and peripheral oedema. Tanezumab treatment did not appear to adversely affect gastrointestinal, cardiovascular, liver or kidney function or have adverse effects on the central nervous system.
As in previous studies, the effect of tanezumab on the peripheral nervous system was rigorously evaluated in this study due to the putative effects of NGF inhibition.3–5 ,7–10 ,21 In the current study, a higher incidence of adverse events of abnormal peripheral sensations was observed in tanezumab-treated versus NSAID-treated subjects. The mechanism for these effects is not known. Most subjects reported transient abnormalities, and low numbers of subjects had clinically relevant neurological examination findings.
The majority of subjects receiving tanezumab having final neurological consultations suggestive of a new or worsening peripheral neuropathy based on diagnostic tests or clinically significant neurological signs were diagnosed with some form of mononeuropathy, predominantly carpal tunnel syndrome; fewer subjects were diagnosed with radiculopathy or polyneuropathy. These results are not the expected pattern for a neurotoxic compound, which typically causes length-dependent polyneuropathy in affected subjects. Previous animal and clinical studies have not indicated any strong evidence of tanezumab neurotoxicity.3–10 ,22 Association of tanezumab with increased numbers of subjects experiencing symptoms of mononeuropathy suggests that NGF inhibition may unmask these conditions.
During conduct of this and other clinical studies of NGF inhibitors in subjects with OA pain, a signal event initially described by investigators as osteonecrosis often leading to TJR raised concerns about joint-related safety of tanezumab and led to a partial clinical hold by the FDA on the entire NGF inhibitor class. Because OA and osteonecrosis have some radiological similarities and can coexist at certain stages, an Adjudication Committee of medical experts in these diseases was formed to examine investigator reports described as osteonecrosis as well as all other reported TJRs not associated with an adverse event of osteonecrosis. In this study, the rate of all-cause TJRs was comparable among the NSAID and tanezumab monotherapy groups and did not increase with increasing tanezumab monotherapy doses, but was twofold greater with tanezumab+NSAID than any of the other groups.
The Adjudication Committee confirmed one investigator report of osteonecrosis in this study; this condition was felt likely to have been present prior to the subject's entry into the study. The remainder of the events reported as osteonecrosis were adjudicated to worsening OA or to another diagnosis. Based on adjudication outcomes, including histopathology confirmation in some cases, it appears that many initial reports of osteonecrosis were based on appearance of fragments of necrotic bone due to bone failure. While commonly seen in end-stage OA and rapidly progressive OA, these bone fragments differ histologically from bone infarction and necrosis that show considerable reparative/reactive changes.
The risk of rapidly progressive OA with tanezumab was greatest when coadministered with NSAIDs and with increasing dose of tanezumab either as monotherapy or combined with NSAIDs (see online supplementary text 5). Both index (identified by the patient as most symptomatic and therefore under evaluation) and non-index joints were affected by rapidly progressive OA. Many subjects entering the study had a history of multiple joint OA disease; thus, our results are not totally surprising. The one patient treated with NSAIDs alone who was adjudicated with rapidly progressive OA had bilateral events in the knees. Across all tanezumab clinical studies, including this study, evidence indicated that OA was present in the affected joint prior to treatment in nearly 90% of subjects affected with rapidly progressive OA. One subject had minimal to no OA in the affected joint; however, the subject experienced a subchondral insufficiency fracture that was evident prior to the determination of rapidly progressive OA. There was no available information to make a determination in the remaining subjects. Finally, a proportion of the affected subjects had either pre-existing rapidly progressive OA prior to study entry, atrophic hip OA or evidence of subchondral insufficiency fracture (affecting primarily the knee) prior to or coincident with the event of rapidly progressive OA.
On 12 March 2012, the FDA Arthritis Advisory Committee reviewed results from this study and the tanezumab programme overall.19 ,23 ,24 Adjudication results presented by the FDA internal and external experts were generally consistent with those provided by the Adjudication Committee assembled by Pfizer.19 ,23 ,24 The committee endorsed continued clinical development of tanezumab and other NGF inhibitors with inclusion of additional measures to minimise risk and further protect patient safety in future studies. On 28 August 2012, the FDA lifted the partial clinical hold on tanezumab, allowing resumption of clinical studies for OA and all other chronic pain conditions.
Based on the thorough assessment of the joint-safety-related outcomes in this study and across all tanezumab clinical studies, the following risk minimisation measures were identified to optimise the benefit–risk profile of tanezumab for continued clinical development of this therapy: (1) exclude chronic concomitant NSAID use, (2) exclude tanezumab doses that have been explored and do not demonstrate benefit over lower doses in the condition under study, (3) exclude subjects with evidence of rapidly progressive OA or risk factors for such from participating in clinical studies and (4) discontinue treatment with study medication in subjects who fail to achieve adequate pain relief. Future studies will be focused on subjects with an inadequate response who cannot tolerate or who have a contraindication for approved or standard of care therapies.
In conclusion, subjects receiving only partial relief of OA pain with an NSAID received greater benefit from treatment with tanezumab. While tanezumab+NSAID combination therapy provided greater relief of OA pain than NSAID treatment alone, no compelling evidence of benefit over tanezumab monotherapy was noted and any small differences in efficacy were negated by greater numbers of subjects experiencing treatment-limiting or irreversible safety outcomes.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figure 1
- Data supplement 3 - Online figure 2
- Data supplement 4 - Online table 1
- Data supplement 5 - Online table 2
- Data supplement 6 - Online table 3
- Data supplement 7 - Online table 4
- Data supplement 8 - Online table 5
Footnotes
Handling editor Tore K Kvien
Subject consent Subjects provided written informed consent before the initiation of protocol-specified procedures.
Contributors All authors have made substantial and equal contributions to the conception and design of the study, acquisition of data, or analysis and interpretation of data; drafting the article or revising it critically for important intellectual content; and final approval of submitted manuscript. Specifically, TJS, EFE and ELHS were involved in acquisition of data; review and interpretation of data; drafting the article or revising it critically for important intellectual content; and final approval of submitted manuscript. HSG was involved in design and monitoring of the study; review, analysis and interpretation of data; revising the manuscript for important intellectual content; and final approval of submitted manuscript. MDS, MTB, CRW and KMV were involved in conception and design of the study; analysis and interpretation of data; revising the manuscript critically for important intellectual content; and final approval of submitted manuscript.
Funding This study was funded by Pfizer Inc. Editorial/medical writing support was provided by Christina McManus of Engage Scientific Solutions and was funded by Pfizer Inc.
Competing interests TJS received consulting fees from Abbott, Merck, Regeneron, Pfizer Inc, Winston Laboratories; performs clinical research for Genzyme, Eli Lilly, Pfizer Inc, Nuvo Research, Nordic Bioscience; and holds equity in NicOx. EFE has received research grants and consulting fees from Novartis, Transdel, Travanti, Bayer and Pfizer Inc, and is a speakers bureau presenter for Pfizer Inc. ELHS has no conflicts of interest to disclose. HSG, MDS, MTB, CRW and KMV are employees of Pfizer Inc.
Patient consent Obtained.
Ethics approval This study was conducted in compliance with the Declaration of Helsinki and all International Conference on Harmonization Good Clinical Practice Guidelines. In addition, all local regulatory requirements were followed; in particular, those affording greater protection to the safety of study participants. The final protocol, any amendments and informed consent documentation were reviewed and approved by independent ethics committees at each participating investigational study centre.
Provenance and peer review Not commissioned; externally peer reviewed.