Article Text

Abstract
Objectives Since clinical non-response to 2×1000 mg rituximab has previously been found to be associated with incomplete B cell depletion, we determined, in a randomised controlled proof of concept study, whether patients with initial incomplete B cell depletion would benefit from an additional infusion of rituximab at week 4.
Methods Patients with active rheumatoid arthritis despite methotrexate received a first infusion of rituximab 1000 mg and were tested for persistent B cells using highly sensitive flow cytometry on day 15. All received a second infusion of 1 g (according to license), but patients with persistent B cells were subsequently randomised double-blind to receive, 2 weeks later, either a third infusion of 1000 mg rituximab or placebo. Clinical response was determined by European League Against Rheumatism (EULAR) and American College of Rheumatology (ACR) criteria.
Results Baseline characteristics were balanced between groups. Treatment with 3×1000 mg rituximab resulted in significantly greater depletion (lower B cell and plasmablast numbers between 8 and 28 weeks) paralleled by significantly better EULAR and ACR20 response rates at 40 weeks (p=0.035 and p=0.027, respectively) and 52 weeks (p=0.021 and p=0.043, respectively) compared with 2×1000 mg. Immunoglobulin titres remained stable in both arms, and adverse event rates were balanced.
Conclusions In rituximab-treated patients with incomplete B cell depletion (predictive of poor response), an extra 1000 mg infusion of rituximab at 4 weeks produced both better depletion and clinical responses than placebo with no worsening of safety. Degree of depletion is an important, but modifiable, determinant of response.
- B cells
- DMARDs (biologic)
- Rheumatoid Arthritis
- Treatment
Statistics from Altmetric.com
Introduction
The licensed dose of two infusions of 1000 mg rituximab in combination with methotrexate (MTX) is effective at reducing disease activity at 6 months in rheumatoid arthritis (RA). However, 35% of patients do not achieve a European League Against Rheumatism (EULAR) moderate response1 and, of those who initially respond, most relapse in the next 6–12 months.2
Patients seronegative for rheumatoid factor and anti-cyclic citrullinated protein (anti-CCP) have worse responses, suggesting that these patients have non-B-cell-mediated disease and require a different therapeutic approach.3 ,4 In the seropositive population, much of the variability in quality and duration of response appears to be related to differences in baseline B lineage cell numbers or degree of B lineage cell depletion. In clinical non-responders, flow cytometric and gene expression studies have shown that baseline numbers of plasmablasts and postswitch memory B cells in blood are increased.5–7 After rituximab, B cell depletion is incomplete if measured using highly sensitive flow cytometry (HSFC) that is able to enumerate the low number of plasmablasts that persist after rituximab.7 ,8 These observations therefore suggest that in seropositive patients failure to respond to rituximab indicates that B cell and plasma cell numbers or activity have been insufficiently inhibited rather than these patients having non-B-cell-mediated disease.
In support of this concept, we have previously shown that patients with low number of plasmablasts at baseline may have complete B cell depletion and good clinical response with a lower dose of rituximab,9 and patients with high number of plasmablasts at baseline, incomplete depletion and clinical non-response may respond well to a second cycle of therapy at 6 months.7
The purpose of the present study was to test the hypothesis that, in patients with incomplete B cell depletion, an extra dose of rituximab at 4 weeks would enhance depletion and clinical response.
Methods
This was a randomised double-blind placebo-controlled trial in which patients who had persistent B cells after a first infusion of rituximab were randomised to a total dose of 2×1000 mg or 3×1000 mg rituximab. The Glasgow West Research Ethics Committee reviewed the study, and all patients provided written informed consent. The study was registered with EUDRACT reference 2006-005640-81.
Patients
Patients were recruited in the Leeds Teaching Hospitals NHS Trust biologics clinic by EMV, SD, MHB and PE between September 2007 and April 2009. Patients meeting 1987 American College of Rheumatology (ACR) criteria for RA were recruited if they had DAS28>3.2 at screening despite stable dose MTX ≥10 mg/week (for at least 4 weeks); an inadequate response to at least one other previous disease-modifying antirheumatic drug (DMARD); and positivity for rheumatoid factor (>40 IU/mL) and/or CCP2 (>7 U/mL).
HSFC was performed 2 weeks after a first infusion of 1×1000 mg rituximab, and patients with detectable naïve or memory B cells or plasmablasts (any subset >0.0001×109/L) were randomised. The week 2 measurement was used to select patients because our previous studies showed this was more predictive of non-response than degree of depletion after both infusions of a licensed dose therapy.8 ,10 ,11
Previous treatment with tumour necrosis factor (TNF) blocking agents or other non-cell-depleting biologics was permitted (but not required) after a minimum washout period of 4 weeks prior to screening. Oral corticosteroids (≤10 mg/day prednisolone or equivalent) were permitted if stable for at least 4 weeks prior to screening and non-steroidal anti-inflammatory drugs (NSAIDs) were permitted if at stable dose for at least 2 weeks prior to screening. Other concomitant DMARDs were not permitted and had to be withdrawn at least 4 weeks prior to rituximab therapy. Exclusion criteria included other autoimmune or inflammatory disease, intra-articular or parenteral corticosteroids in the last 4 weeks, previous rituximab, active infection, previous malignancy, low IgG or positivity for hepatitis B or C.
Treatment
Patients were randomised to either one further infusion of 1000 mg at week 2 and one infusion of placebo at week 4 (a total of 2×1000 mg rituximab) or two further infusions of 1000 mg rituximab at weeks 2 and 4 (a total of 3×1000 mg rituximab). One hundred milligrams of methylprednisolone was administered prior to the first two infusions of rituximab only so that patients in each arm received a total of 2×100 mg methylprednisolone. Randomisation was performed 1:1 in blocks of four patients by a code generated before the study. Randomisation was performed by the trial pharmacist, and patients and investigators were blinded to treatment allocation for the duration of the study.
MTX was continued at the dose taken at baseline except for safety reasons. Continued use of NSAIDs taken at baseline was permitted and only adjusted in case of adverse event. Additional corticosteroid use was allowed as follows: up to two courses of 40 mg prednisolone for up to 2 weeks were allowed for non-RA conditions. Intramuscular or intra-articular corticosteroids for RA were permitted in exceptional cases after discussion with investigators but not within 4 weeks of screening or primary or secondary outcome assessments.
After week 28, patients were permitted to receive further cycles of rituximab in standard dose (2×1000 mg) if they had clinical relapse with a minimum DAS28 increase of 0.6 since the prior assessment. Such patients were considered as EULAR and ACR20 non-responders at subsequent assessments. Investigators and patients remained blinded to treatment allocation at assessment of relapse, re-treatment and subsequent assessments.
Assessments
Tender and swollen joint counts in 28 joints performed by a single specialist nurse, patient visual analogue scale for global health, disease activity and pain, physician global visual analogue scale, health assessment questionnaire disability index, erythrocyte sedimentation rate, C-reactive protein (CRP), rheumatoid factor, immunoglobulins and routine laboratory assessments were performed at the following time points: 0, 8, 12, 16, 28, 40 and 52 weeks.
The primary efficacy outcome was the proportion of patients achieving ACR20 at week 28. Secondary endpoints included EULAR and ACR20, ACR50 and ACR70 responses at 14, 28, 40 and 52 weeks; B cell naïve, memory and plasmablast subsets at the same time points, immunoglobulin titres and adverse event rates.
Flow cytometry
Peripheral blood B cell subsets were measured using HSFC as previously described at baseline after each infusion of rituximab, then 3-monthly. Briefly, B cell numbers and subsets were enumerated following standard cell surface staining techniques using a sequential gating strategy with six colour flow cytometry, counting 500 000 events. First B cells were identified using CD19 PerCP-Cy5.5 (BD Biosciences, Oxford, UK) and CD38 PE-Cy7 (BD Biosciences, Oxford, UK) expression and light scatter characteristics to identify B cells. CD3 FITC and CD14 PE (BD Biosciences) were used to exclude contaminating events, and B cells were classified according to expression of CD38 PE-Cy7 and CD27 APC (BD Biosciences) as naïve (CD19++CD27−), memory (CD19++CD27+) and preplasma (CD19+/−CD27++CD38++). CD45 APC-Cy7 (BD Biosciences) was used to identify total leucocytes for calculation of absolute B cell subset numbers.
All screened patients met inclusion and exclusion criteria at the time of collection of baseline data and for 4 weeks prior to baseline. However, (i) in seven patients, MTX dose was only 7.5 mg at screening; (ii) five patients had received oral or intramuscular steroids within 4 weeks prior to screening; and (iii) one patient had active infection within 2 weeks of screening. Of these patients, one with MTX dose 7.5 mg (for 3 years, due to toxicity) and two who had received additional corticosteroids prior to screening were randomised (all to 3×1000 mg). The others were either withdrawn prior to baseline or not randomised due to complete B cell depletion at week 2. The data for these patients have been included in the analysis. These deviations from protocol were reported to the Medicines and Healthcare Products Regulatory Agency.
This study was closed early by the investigators (during safety follow-up, but after collection of the above primary and secondary endpoint data in all patients) for logistical reasons. Efficacy and safety outcomes have therefore been reported for 12 months follow-up from baseline.
Statistical analysis
As a proof of concept study, no formal power calculation was performed. Descriptive statistics were provided as proportions for response data, mean and 95% CI for normal distributed interval data and medians and IQR for non-normally distributed or ordinal data. Changes in normally distributed continuous outcome data were compared using t tests, and changes in non-normally distributed or ordinal outcome data were compared using Wilcoxon rank-sum test. Response rates between groups were compared using univariate binary logistic regression.
Results
Twenty-five patients with initial incomplete depletion who satisfied inclusion and exclusion criteria were enrolled, randomised and analysed for the primary outcome measure (13 in the 2×2000 mg group and 12 in the 3×1000 mg group). In order to recruit these 25 patients, 60 patients were treated with rituximab. The rate of incomplete initial depletion was therefore 42%.
Baseline characteristics
Baseline disease characteristics are shown in table 1. These were typical for an active RA population and balanced between treatment arms; although there was a trend to higher median CRP in the 2×1000 mg group, DAS28 was similar in each group. In total, 54% and 92% of patients in the 2×1000 mg and 3×1000 mg groups respectively were biologic-naïve. Only one patient had received multiple TNF inhibitors. None of the patients recruited had received non-TNF biologics.
Baseline characteristics
B cell depletion and repopulation
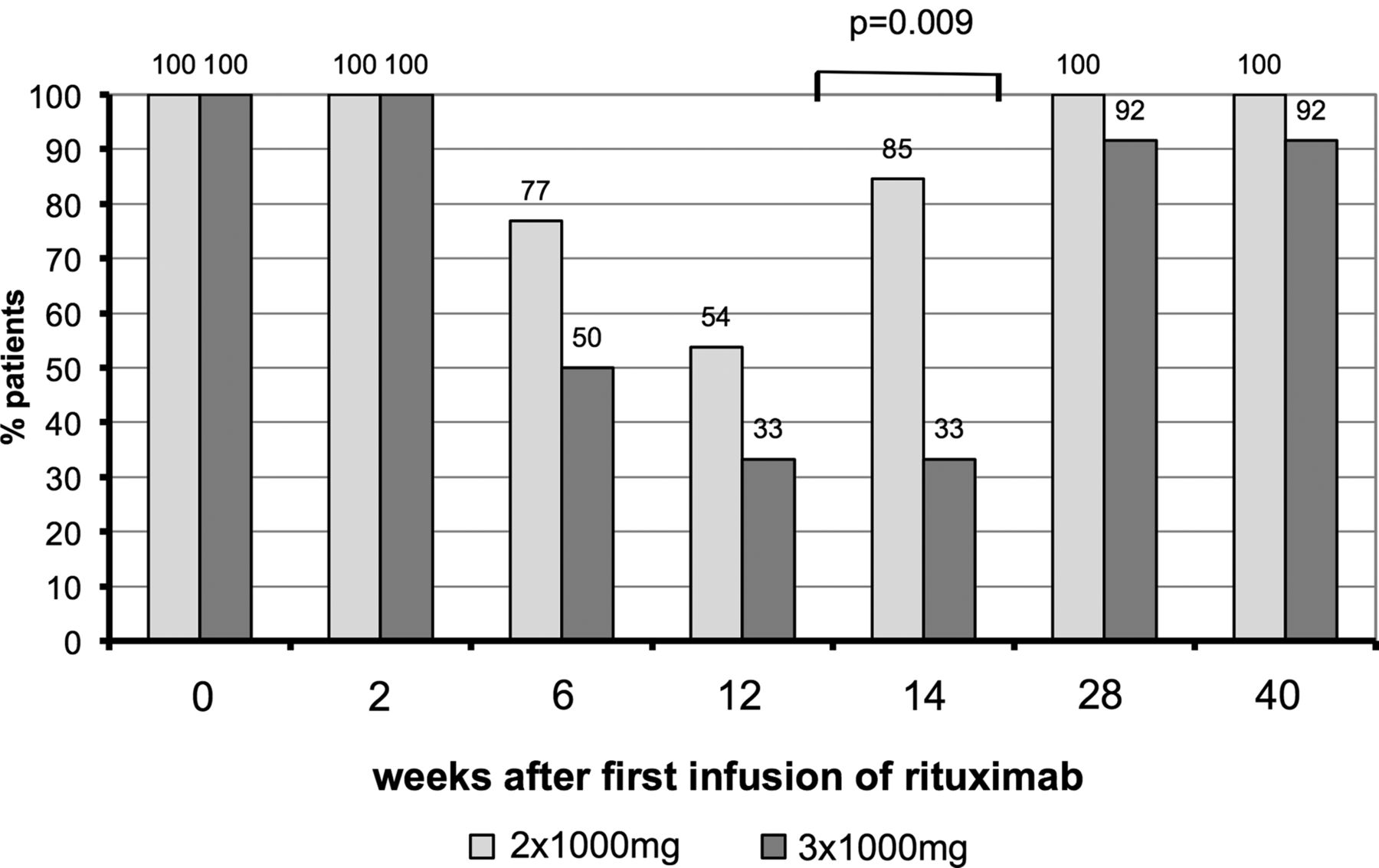
The number of each B cell subset and total B lineage cells is shown in figure 1. Median cell count was lower for each subset at time points from 8 to 28 weeks in patients who received the 3×1000 mg dose compared with the 2×1000 mg dose. These differences were significant for total B cells at 16 and 28 weeks and for plasmablasts at 8 and 16 weeks. The proportions of patients in whom B cells of any subset were detectable are shown in figure 2. Significantly more patients had detectable B cells in the 2×1000 mg group at week 16 (85% vs 33%, p=0.009).
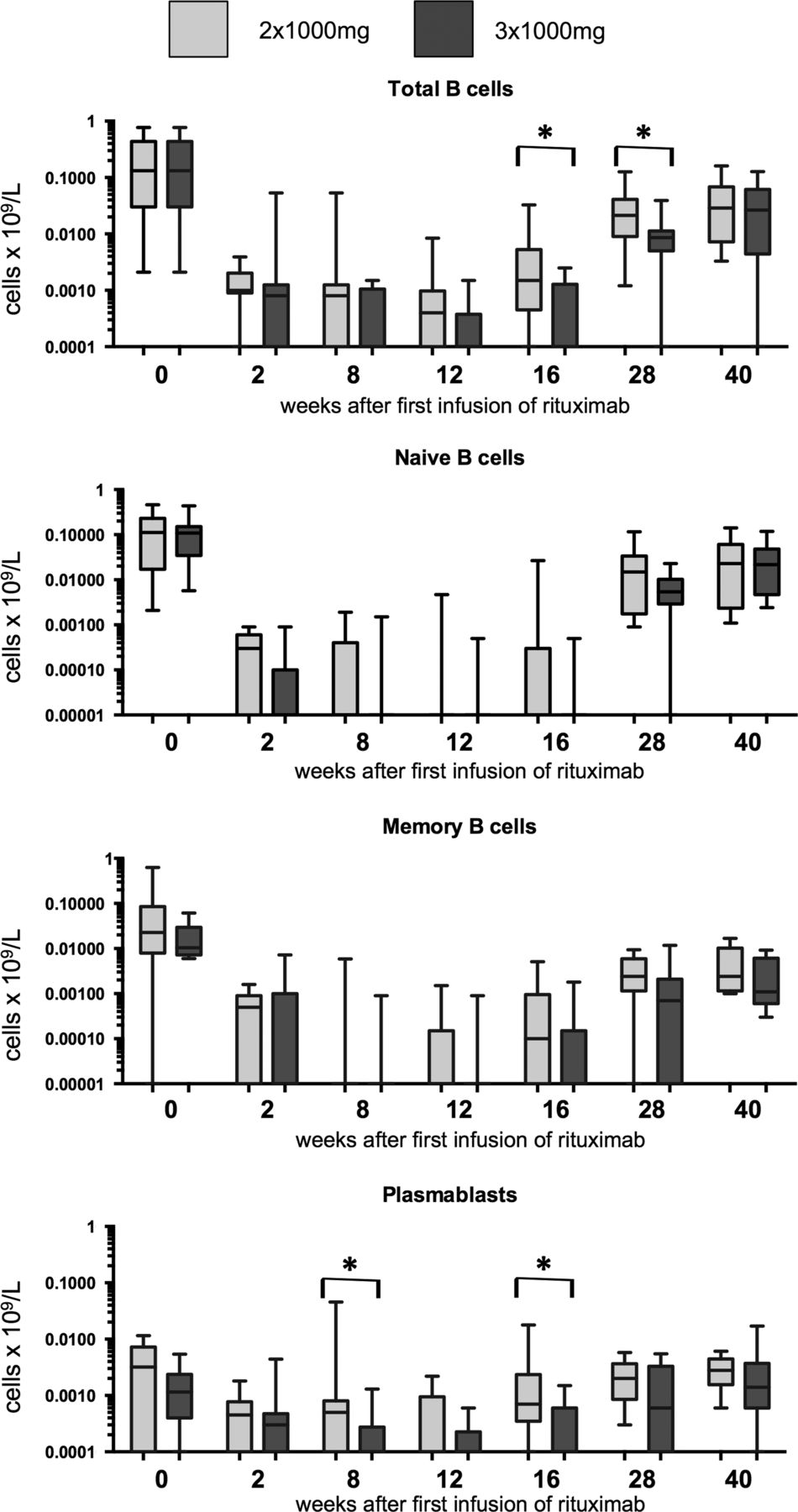
B cell subsets as measured by highly sensitive flow cytometry. B cell subsets were defined as naïve (CD19+CD27−), memory (CD19+CD27+CD38−) and plasmablast (CD19+/−CD27++CD38++). The lower limit of detection was 0.0001×109 cells/L for each subset individually. Total B cells were calculated as the sum of all three subsets. Boxes represent 1st and 3rd quartiles, and the horizontal line with each box represents the median. Whiskers show the 95% CI. Brackets with asterisks indicate that significantly different results between treatment groups with p<0.05 using Wilcoxon rank-sum test.

Proportion of patients with detectable B cells of any lineage. Brackets with p values indicate results of univariate binary logistic regression comparing B cell status for each group.
Because a weak trend to lower plasmablast levels in the 3×1000 mg group (p=0.347) was observed at baseline, we also tested within-group change in plasmablasts for each rituximab dose. After the first 1000 mg infusion of rituximab, a significant reduction in plasmablasts was observed in each group as expected (p=0.012 and p=0.042 for 2×1000 mg and 3×1000 mg, respectively).
However, after randomisation at 2 weeks, further reduction was only observed in the 3×1000 mg group. In the 2×1000 mg group, there was no substantive or significant change in plasmablasts between 2 and 8 weeks (p=1.0). In contrast, in the 3×1000 mg group, there was a trend to further reduction (p=0.212).
Hence, overall between weeks 0 and 8 a significant reduction in plasmablasts was observed for 3×1000 mg (p=0.009) but only a smaller and non-significant reduction for 2×1000 mg (p=0.135).
Within -group plasmablast depletion has been illustrated separately in figure 3.

Details of plasmablast depletion within each group. The first infusion was given at week 0. The next one or two infusions were given at weeks 2 and 4. Values shown are log10 ((value+0.0001)×10 000). Boxes represent 1st and 3rd quartiles, and the horizontal line with each box represents the median. Whiskers show the 95% CI. p Values refer to within-group Wilcoxon signed rank tests. R indicates an infusion of 1×1000 mg rituximab.
Efficacy
Between 28 and 40 weeks, four patients relapsed and were re-treated (three in the 2×1000 mg group and one in the 3×1000 mg group). Between 40 and 52 weeks, a further seven patients relapsed and were re-treated (four in the 2×1000 mg group and three in the 3×1000 mg group). These patients were classified as ACR and EULAR non-responders at subsequent time points.
At 28 weeks, ACR20 and EULAR moderate or good response rates were numerically, but not statistically, superior in the 3×1000 mg group. Also, 8/13 (62%) and 8/12 (67%) of patients in the 2×1000 mg and 3×1000 mg groups respectively achieved ACR20 responses and 10/13 (77%) and 11/12 (92%) of patients achieved EULAR moderate or good responses, respectively. After 28 weeks, response rates worsened in the 2×1000 mg group, with greater number of patients relapsing, and were maintained in the 3×1000 mg group so that a significant difference between groups was observed (figure 4). At 40 weeks, 4/13 (31%) and 9/12 (75%) of 2×1000 mg and 3×1000 mg patients respectively achieved ACR20 responses (p=0.027) and 7/13 (54%) and 11/12 (92%) achieved EULAR moderate or good responses, respectively (p=0.035). One patient in each arm did not attend for the 52-week assessment. At 52 weeks, 1/12 (8%) and 5/11 (45%) of 2×1000 mg and 3×1000 mg patients respectively achieved ACR20 responses (p=0.043) and 2/12 (17%) and 7/11 (64%) achieved EULAR moderate or good responses, respectively (p=0.021). Differences in ACR50, ACR70 and EULAR good response rates were not significantly different, but there was a trend for difference in ACR70 response rate at 52 weeks, which was achieved by 0% and 25% of patients in the 2×1000 mg and 3×1000 mg groups, respectively (p=0.052). Full data on all outcome measures are shown in the online supplementary material.

Clinical responses for key outcome measures. Brackets with p values indicate results of univariate binary logistic regression comparing response rate for each group.
We compared B cell numbers in patients with an initial EULAR moderate or good response at week 28 who still had a moderate or good response at week 40 (n=17, ‘maintained response’) with patients who had EULAR non-response or who had required re-treatment by week 40 (n=4, ‘lost response’). Significantly lower total B cell counts at weeks 6 (p=0.042), 14 (p=0.025) and 28 (p=0.003) were observed in patients who maintained response. B cell counts were not associated with maintenance of response at 52 weeks.
Safety
At 6 months, all patients had received only the initial randomised cycle of rituximab. There were a total of 38 adverse events in the 2×1000 mg group and 40 adverse events in the 3×1000 mg group. Of these, 9 and 12 (for the 2×1000 mg and 3×1000 mg groups, respectively) were infective and 7 and 5 were arthritis-related.
By 12 months, some patients had also received an additional cycle of standard-dose rituximab as described above. Safety data for 12 months have been summarised, including the re-treated patients. There were a total of 54 and 59 adverse events in the 2×1000 mg and 3×1000 mg groups, respectively. In total, 13 and 17 were infective and 11 and 11 were arthritis-related. There were five serious adverse events. Of these, four were hospitalisations for elective surgery, which was uncomplicated (one and four in the 2×1000 mg and 3×1000 mg groups, respectively). One was hospitalisation for an infected foot ulcer, which resolved without sequelae (3×1000 mg group).
A significant decrease in IgM (p=0.001) and IgG (p=0.029) but not IgA (p=0.293) titres was observed at 26 weeks (figure 5). By comparing either change or percentage change in each immunoglobulin class between treatment arms, no significant difference was observed, but there was a trend to greater percentage reduction in IgG in patients who received 3×1000 mg rituximab. Mean (95% CI) percentage change was 3.4% (−7.5–14.3%) for 2×1000 mg and 14.4% (2.7–26.2%) (p=0.133). No patient developed IgG below the lower limit of normal.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Change in titres of total serum immunoglobulin IgA, IgG and IgM. Points indicate the mean for each group, and error bars indicate 95% CIs. Horizontal dotted lines indicate the laboratory upper and lower limits of normal.
Total rituximab use over 12 months
Using our protocol of re-treatment of initial non-responders with 2×1000 mg at 6 months, and re-treatment of initial responders with 2×1000 mg on clinical relapse, the mean total rituximab dose administered in the first 12 months was 3.83 g per patient in the 2×1000 mg arm and 3.90 g per patient in the 3×1000 mg arm.
Discussion
In this study, we found that, in patients with a biomarker predicting worse clinical response, B cell depletion and clinical response could be enhanced by an increased dose of rituximab. Although no difference in clinical response was observed at our primary endpoint at 28 weeks, responses were longer lasting in the 3×1000 mg arm, with high rates of loss of response and relapse between 28 and 52 weeks in patients with incomplete depletion treated with 2×1000 mg. These findings indicate that (i) the current licensed dose of rituximab may not be the most effective for all patients and that (ii) monitoring B cells with HSFC may allow targeting of patients for more intensive therapy.
Previous research has largely focused on the use of lower rituximab doses. In early RA with non-radiographic endpoints, outcomes were equivalent using 2×500 mg rituximab to the licensed dose.12 However, in early RA, radiographic outcome was better using 2×1000 mg13 and in established RA EULAR response was better using 2×1000 mg.14 We have previously shown that the efficacy of lower doses of rituximab is dependent on initial B cell depletion.11 Recent data have indicated that 1×1000 mg may be similarly effective to 2×1000 mg in a repeat cycle.15 In this study, we extend this evidence by demonstrating the ability to overcome incomplete depletion using higher initial doses of rituximab. By tailoring rituximab doses to B cell subsets and depletion, the most appropriate long-term management may therefore be to use different doses for response induction and maintenance, with a larger initial dose of rituximab in some patients, then smaller doses in repeat cycles.
In this study, we chose to only randomise patients who had incomplete depletion of B cells at 2 weeks. This was because of the potential for increased rates of hypogammaglobulinaemia and infection using inappropriately high-dose rituximab and our previous data demonstrating consistently good responses using standard dose in patients with complete depletion. We therefore did not feel that investigation of higher dose therapy was justified in patients with complete depletion at standard dose. Wider implementation of this and other strategies that adjust doses to individual patients’ needs would therefore require the use of HSFC in routine clinical practice. Experience in haematology has demonstrated that it is feasible and may be cost effective to stratify therapy according to similar flow cytometry techniques. Treating to maximal response by minimal residual disease flow cytometry has been associated with better clinical outcomes in chronic lymphocytic leukaemia in a study in our institution,16 and the efficacy of this strategy was subsequently confirmed in a multicentre trial.17 Minimal residual disease flow cytometry is under consideration by the Food and Drug Administration as a surrogate endpoint for clinical trials in chronic lymphocytic leukaemia.18 While optimal treatment algorithms are still emerging, in our centre, HSFC is now routinely used in rituximab therapy to guide re-treatment decisions, dose adjustment and biological switching.
Clinical responses at 28 weeks were better than in our previous study in both groups. This is likely to be due to differences in baseline disease activity and prior therapies. A more significant difference between treatment groups may have been seen at 6 months in a more severe disease group. Nevertheless, this study included a challenging resistant RA population with intensive assessments prior to baseline (patients underwent multiple imaging modalities as part of substudies that will be published separately). This is likely to have contributed to the deviations from protocol inclusion and exclusion criteria detailed in the ‘Methods’ section. We do not believe that these deviations influenced the conclusions of the study. Future trials in this population should allow flexibility in entry criteria in order to ensure they are representative of ‘real life’ resistant RA.
In this trial, we only assessed seropositive patients. In our centre, seronegative patients were not treated with rituximab due to previous evidence demonstrating inferior efficacy in this group. It is possible that effect of dose may be important in the poorly responding seronegative group, but this would require a further trial. Similarly, assessment of effect of higher dose rituximab in patients with good initial depletion would also require a further trial.
Although higher dose might incur additional initial drug cost, the protocol in this study was cost-neutral over a 12-month period using on-demand re-treatment. The increased cost of initial treatment was offset by an extension of time to re-treatment, and with the advantage of a longer period of stable disease between cycles of rituximab. Given that repeat cycles were given later in the 3×1000 mg arm, this strategy may become even more cost effective with further follow-up and repeat cycles. The best long-term re-treatment strategy with rituximab has not yet been established.19 Some physicians therefore use re-treatment at a fixed 6-monthly interval. By comparing trials that used on-demand re-treatment with other trials that used 6-monthly re-treatment, efficacy appeared better with 6-monthly treatment,20 although the fixed re-treatment trials recruited milder and earlier disease that may have biased those groups of patients to better responses. The benefits of the 3×1000 mg dose may not be apparent in comparison to regimens that re-treat all patients every 6 months. However, multiple cycles of rituximab increase the risk of hypogammaglobulinaemia and serious infection as well as cost, so protocols that achieve stable disease control without excessive therapy are desirable.
Clinical trials testing the duration of response to higher dose rituximab and the ability of early depletion to select patients most likely to benefit should be conducted and could be designed and powered based on our results.
Acknowledgments
The authors thank Christine Thomas, Matthew Robinson, Beverley Wells, Domini Bryer, Sarah Fahy, Steve Rose and Anne-Maree Keenan for their assistance in coordination of this study.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
Handling editor Tore K Kvien
Contributors All authors contributed to the conception and design, or analysis and interpretation of data. All authors also contributed to drafting the article or revising it critically for important intellectual content and approved the final version to be published.
Funding National Institute of Health Research and Roche.
Competing interests EMW was funded by a NIHR Research Training Fellowship (grant number RTF/01/097) and is a NIHR Clinical Lecturer. MHB is a NIHR Clinician Scientist. PE is Arthritis Research UK Professor of Rheumatology. Roche supplied rituximab and a research grant for this study.EMV, SD, MHB and PE received honoraria and research grants from Roche. ACR has received research grant support from Becton Dickinson.
Ethics approval Glasgow West Research Ethics Committee.
Provenance and peer review Not commissioned; externally peer reviewed.