Article Text

Abstract
Objectives: To identify the main causes of morbidity and mortality in patients with antiphospholipid syndrome (APS) during a 5-year period and to determine clinical and immunological parameters with prognostic significance.
Methods: The clinical and immunological features of a cohort of 1000 patients with APS from 13 European countries who had been followed up from 1999 to 2004 were analysed.
Results: 200 (20%) patients developed APS-related manifestations during the 5-year study period. Recurrent thrombotic events appeared in 166 (16.6%) patients and the most common were strokes (2.4% of the total cohort), transient ischaemic attacks (2.3%), deep vein thromboses (2.1%) and pulmonary embolism (2.1%). When the thrombotic events occurred, 90 patients were receiving oral anticoagulants and 49 were using aspirin. 31/420 (7.4%) patients receiving oral anticoagulants presented with haemorrhage. 3/121 (2.5%) women with only obstetric APS manifestations at the start of the study developed a new thrombotic event. A total of 77 women (9.4% of the female patients) had one or more pregnancies and 63 (81.8% of pregnant patients) had one or more live births. The most common fetal complications were early pregnancy loss (17.1% of pregnancies) and premature birth (35% of live births). 53 (5.3% of the total cohort) patients died. The most common causes of death were bacterial infection (21% of deaths), myocardial infarction (19%) and stroke (13%). No clinical or immunological predictor of thrombotic events, pregnancy morbidity or mortality was detected.
Conclusion: Patients with APS still develop significant morbidity and mortality despite current treatment (oral anticoagulants or antiaggregants, or both).
Statistics from Altmetric.com
The antiphospholipid syndrome (APS) is a systemic autoimmune disorder characterised by a combination of arterial and/or venous thrombosis, pregnancy morbidity and raised titres of antiphospholipid antibodies (aPL).1 To describe the course of the disease in patients with APS, we started in 1999 a multicentre observational study of 1000 European patients with a mean age of 42 years. The clinical and immunological characteristics of these patients when entered in the study have been previously reported.2 Since then, this cohort of patients has been followed up by the same doctors during the ensuing 5 years.
The aims of this study were to identify the main causes of morbidity and mortality during this period (1999–2004) and to determine clinical and immunological parameters with prognostic significance in a cohort of patients with APS.
Patients and methods
Patient selection
The study (“Euro-Phospholipid” project) started in 1999 with a multicentre and consecutive design. To gather a sizeable series of patients, 20 tertiary referral university centres, with substantial experience in the management of patients with APS, from 13 countries (Belgium, Bulgaria, Denmark, France, Germany, Greece, Hungary, Israel, Italy, the Netherlands, Portugal, Spain and United Kingdom) agreed to take part in the study. The final cohort included 1000 unselected patients who met the proposed preliminary criteria for the classification of definite APS.3
All patients have been followed up by the same doctors during the ensuing 5 years (1999–2004) with regular visits to the outpatient clinics at least every 3–6 months, depending on the severity of the disease and admitted to hospital if necessary. One hundred and fifty-one (15.1%) patients were lost to follow-up. The observation period stopped in 2004 or at the time of the latest patient information if lost to follow-up or at death. The study was performed according to the principles of the Declaration of Helsinki.
Definition of clinical features
To minimise possible interobserver bias and to monitor the accuracy of the data collection, a standard protocol was used and the variables of this protocol were carefully discussed by all the participating doctors on several occasions. These variables included causes of morbidity (APS manifestations and other associated medical problems), causes of death and survival. The main APS clinical manifestations evaluated in this prospective study have been previously described.2 Patients were considered to have these manifestations if the diagnosis was firmly confirmed according to the established criteria for each manifestation using laboratory, imaging or Doppler studies or histopathology, with the exception of superficial venous thrombosis and other dermatological features that could be diagnosed on clinical grounds. For histopathological confirmation of thrombosis, no significant evidence of inflammation should be present in the vessel wall. Briefly, among the major clinical manifestations, deep vein thrombosis was confirmed by Doppler studies and/or phlebography, peripheral arterial thrombosis by arteriography, cerebrovascular accident, multi-infarct dementia, cerebral venous thrombosis and transverse myelopathy by computed tomography (CT) and/or magnetic resonance imaging (MRI) scans, pulmonary embolism was confirmed by ventilation/perfusion pulmonary scintigraphy, myocardial infarction by raised cardiac enzymes and electrocardiogram and intra-abdominal infarctions by CT and/or MRI scans. Other APS-related manifestations, but not considered as classification criteria in the international consensus statement on an update of the classification criteria for definitive APS,4 were thrombocytopenia (<100×109/l, confirmed at least twice 12 weeks apart), livedo reticularis, epilepsy, skin ulcers and heart valve lesions (all diagnosed according to the definitions of this consensus statement).4 Patients were considered to have catastrophic APS if they presented with an acutely devastating APS with multiple (>3) organ involvement, mainly affecting small vessels supplying organs and presenting over a short period of time (<1 week), as previously defined.5
Pregnancy morbidity was considered when the following established definitions were fulfilled: (a) one or more unexplained deaths of a morphologically normal fetus at or beyond the 10th week of gestation, with normal fetal morphology documented by ultrasound or by direct examination of the fetus, or (b) one or more premature births of a morphologically normal neonate before the 34th week of gestation because of eclampsia or severe pre-eclampsia defined according to standard definitions, or recognised features of placental insufficiency, or (c) three or more unexplained consecutive spontaneous abortions before the 10th week of gestation, with maternal anatomical or hormonal abnormalities and paternal and maternal chromosomal causes excluded.4
The main clinical manifestations related to systemic lupus erythematosus (SLE) evaluated in this study were defined according to the American Rheumatism Association glossary committee6 and are described in detail elsewhere.7 Other vascular risk factors evaluated in this study included diabetes mellitus (any previous diagnosis of diabetes or two glycaemic controls of ⩾126 mg/dl), hypertension (blood pressure ⩾140/90 mm Hg), hypercholesterolaemia (total cholesterol blood levels ⩾200 mg/dl, high-density lipoprotein cholesterol ⩽35 mg/dl and or low-density lipoprotein cholesterol ⩾160 mg/dl) and smoking (⩾10 cigarettes/day). Diagnoses of the other associated medical problems that appeared during the study (infections, malignancies, haemorrhages, drug side effects, etc) were performed on clinical grounds and confirmed by appropriate complementary techniques. The causes of death were based on information obtained from the clinicians in charge, autopsy reports and/or death certificates.
Laboratory studies
The anticardiolipin antibodies (aCL) of the IgG and IgM isotypes were measured by a β2 glycoprotein I (β2GPI)-dependent enzyme-linked immunosorbent assay (ELISA).8 They were considered positive if present in medium to high titre (>40 GPL or >40 MPL) on two or more occasions, at least 6 weeks apart. Lupus anticoagulant (LA) activity was detected by coagulation assays according to the guidelines of the International Society on Thrombosis and Haemostasis (Scientific Subcommittee on Lupus Anticoagulants/Phospholipid-Dependent Antibodies).9
Statistical analysis
The following independent variables were included in the statistical analysis for the detection of possible predictors of the outcomes of interest (ie, thrombotic events, pregnancy morbidity and mortality): primary APS, SLE, gender, age at onset of APS, comorbidities (diabetes mellitus, hypertension, hypercholesterolaemia and smoking) and aPL (IgG aCL, IgM aCL and LA). Univariate and multivariate Cox proportional hazard regression tests were used to determine associations of our outcomes of interest (dependent variables) with the different independent variables. When several independent variables appeared to have statistical significance in the univariate analysis, they were included in the multivariate analysis. Results of the analysis of continuous variables are indicated as mean (SD). Survival time was defined as the interval from the time the patient entered in the study until death or last contact. Survival probabilities were calculated according to Kaplan–Meier lifetime analysis method. Statistical significance was defined as a p value ⩽0.05.
Results
General characteristics at the beginning of the prospective study
The entire cohort consisted of 820 (82.0%) female and 180 (18.0%) male patients. There were 985 (98.5%) whites, 5 (0.5%) blacks and 10 (1.0%) patients of other races. Mean (SD) age at study entry was 42 (14) years (range 0–82; median, 40). A total of 53.1% had primary APS, 36.2% had APS associated with SLE, 5.0% associated with lupus-like syndrome and 5.7% associated with other diseases. Six patients diagnosed at entry as having a primary APS developed anti-dsDNA antibodies during the 5-year study period and were reclassified as lupus-like syndrome. The main clinical manifestations at the onset of the disease, the cumulative clinical manifestations from the onset until the beginning of the study and the immunological findings when the patients entered in the study have been reported in detail elsewhere.2
APS manifestations and treatment during the study period
Two hundred (20%) patients developed APS-related manifestations during the 5-year study period. Recurrent thrombotic events appeared in 166 (16.6%) patients and the most common were strokes (2.4% of the total cohort), transient ischaemic attacks (2.3%), deep vein thromboses (2.1%) and pulmonary embolism (2.1%). Three out of 121 (2.5%) women with only obstetric APS manifestations at the beginning of the study developed a new thrombotic event during the 5-year study period (one deep vein thrombosis, one stroke and one catastrophic APS, respectively) and only one of these patients was receiving primary thromboprophylaxis with aspirin before the thrombotic event. Nine (0.9%) patients developed an episode of catastrophic APS. Other APS-related manifestations (but not considered as classification criteria) that appeared during the study period were thrombocytopenia (3.7%), livedo reticularis (2.7%), epilepsy (1.7%), skin ulcers (1.7%) and valve thickening/dysfunction (1.7%) (table 1). After performing the multivariate analysis, no statistical differences were detected in the occurrence of APS manifestations depending on the underlying autoimmune disease (primary APS or SLE), the gender, the age at onset of APS or the presence of comorbidities (diabetes mellitus, hypertension, hypercholesterolaemia and smoking). Neither individual aPL (IgG aCL, IgM aCL or LA) nor the combination of some of them was associated with an increased incidence of any specific clinical manifestation.
Main clinical manifestations related to the antiphospholipid syndrome (APS) that appeared during the 5-year follow-up (1999–2004) of the total cohort of 1000 patients
Oral anticoagulants were used in 420 (42%) patients and low-dose aspirin in 350 (35%). When the recurrent APS-related thrombotic events appeared, 90 patients were receiving oral anticoagulants (69 at a target international normalised ratio (INR) of 2–3 and 21 at a target INR >3), 49 were taking aspirin and 27 were neither anticoagulated nor antiaggregated. Thirty-one patients of 420 receiving oral anticoagulants (7.4%) developed haemorrhages (cutaneous in 18 patients, cerebral in 7, gastrointestinal in 4 and intra-abdominal in 2).
A total of 77 women (9.4% of female patients) had one or more pregnancies (range 1–4, total number of pregnancies 105) and 63/77 (81.8%) succeeded in having one or more live births (range 1–3, total number of live births 80). The most common fetal complications were early pregnancy loss (17.1% of pregnancies), late pregnancy loss (6.7% of pregnancies), premature birth (35% of live births) and intrauterine growth restriction (13.7% of live births).
Mortality and causes of death during the study period
During the study period, 53 (5.3%) patients died (21 in the first year, 12 in the second, 10 in the third, 5 in the fourth and 5 in the fifth). They included 38 female (72%) and 15 male (28%). Mean (SD) age at death was 53 (14) years (range 19–79).
The most common causes of death were bacterial infections (20.8% of deaths), myocardial infarction (18.9%), stroke (13.2%), haemorrhage (11.3%), malignancy (11.3%), catastrophic APS (9.4%) and pulmonary embolism (9.4%) (table 2). After performing the multivariate analysis, no statistical differences were detected in the causes of death depending on the underlying disease (primary APS or SLE) or the treatment that patients were receiving (immunosuppressive or anticoagulant agents). A survival probability of 94% was found at 5 years from the time of entry into the study (fig 1). There were no differences in the survival probability depending on the underlying autoimmune disease (primary APS or SLE), the gender, the age at onset of APS, the clinical manifestations, the immunological parameters or the treatment that patients were receiving (immunosuppressive or anticoagulant agents).

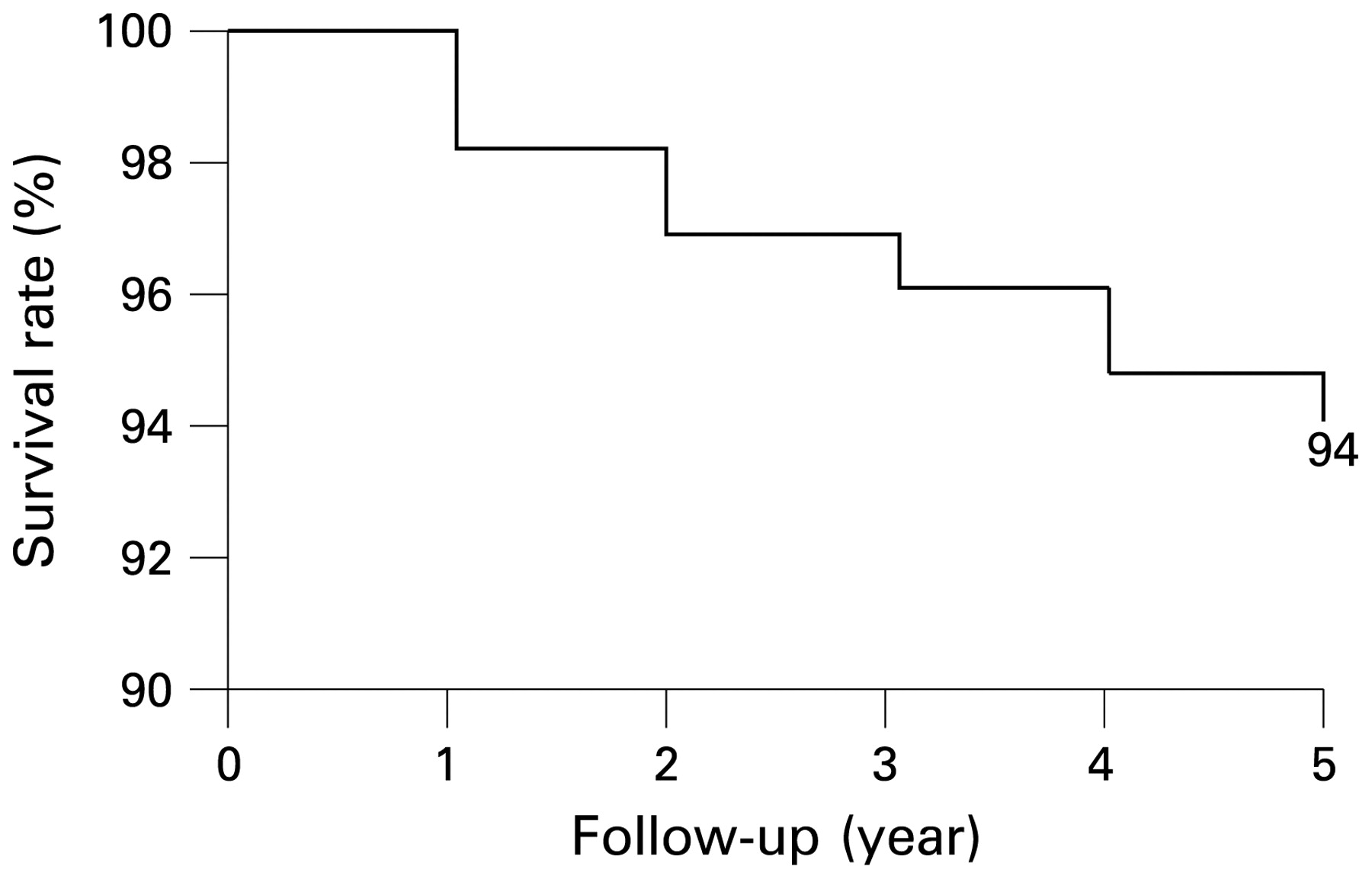{kind=link}
Kaplan–Meier survival curve of the total cohort showing a 94% probability of remaining alive at 5 years from the time of entry into the study.
Causes of death during the 5-year follow-up (1999–2004) of the total cohort of 1000 patients
Discussion
In this study we have described the main APS clinical manifestations as well as the mortality rate and the causes of death in a large cohort of European patients followed up during a 5-year-period (1999–2004). Furthermore, we have attempted to determine possible predictors of several outcomes of interest in the APS. As distinct from previous European epidemiological studies that included patients from one single country,10,11,12,13,14 this study covers a more representative European APS population, including patients from northern, western, southern, central and eastern Europe. The problem of a potential difference in medical care in the participating hospitals has been overcome by careful selection of tertiary referral university centres having clinicians with considerable experience in the management of patients with APS and by careful discussion of the definition of all the outcome variables. Although 151 (15.1%) patients were lost to follow-up, this accounted for only 3% a year and the appearance or absence of the different outcome variables during the period of time that these patients participated in the study was also registered.
We have found a much lower incidence of thrombotic manifestations during this study, compared with the cumulative clinical manifestations before the start of the study (median previous period of evolution, 6 years).2 For instance, the frequency of deep vein thrombosis during this 5-year period was 2.1% while we had previously found a cumulative frequency of 38.9% when the patients entered into the study.2 Additionally, several APS-related manifestations not included in the classification criteria were similarly uncommon—namely, thrombocytopenia (3.7%), livedo reticularis (2.7%), epilepsy (1.7%), skin ulcers (1.7%) and valve thickening/dysfunction (1.7%). Interestingly, strokes and transient ischaemic attacks were the most common recurrent thrombotic events—as they appeared in 2.4% and 2.3% of the total cohort, respectively—while deep vein thromboses were the most common thrombotic events at the study entry. As most of these patients were receiving oral anticoagulants at a target INR between 2 and 3, this might indicate that this treatment mainly protects against venous thrombosis but is not sufficiently protective against arterial thrombosis.
Although the majority of patients in this cohort were receiving either anticoagulant or antiaggregant agents, 230 (23.0%) were not receiving any of these drugs during this study period. This may reflect the “real-world” situation, as many doctors are still reluctant to prescribe any drug for primary thromboprophylaxis to those female patients with APS who had experienced pregnancy morbidity or for long-term secondary thromboprophylaxis several years after the APS thrombotic event. However, 90 patients presented with a recurrent thrombotic event despite anticoagulation (21 of them at a target INR >3). This may reflect a selection bias (patients with more severe clinical manifestations may be more consistently given oral anticoagulants by their doctors), but we believe that this was probably because many patients with APS cannot keep within their target INR.15 Therefore, the INR at the time of the event would have much more value than the target INR but this is difficult to determine in most patients and these data were not consistently obtained in the study. On the other hand, 31/420 patients receiving oral anticoagulants (7.4%) presented with haemorrhages, 13 of them in internal organs (cerebral in seven, gastrointestinal in four and intra-abdominal in two) and in six of them, they were the main cause of death.
In this cohort, low-dose aspirin did not prevent thrombosis in 49/350 (14%) patients who were using it. Erkan et al16 have recently reported the results of a short trial comparing low-dose aspirin with placebo in asymptomatic, persistently aPL-positive subjects. They found a low overall annual incidence rate of acute thrombosis, but a few patients developed vascular events when additional thrombotic risk factors were present and the use of low-dose aspirin did not add any benefit. Although the low-risk profile of the study group in this trial precludes a generalisation of the results to other settings, taken together with the findings in our cohort, it seems that additional oral treatments (eg, the routine use of antimalarial drugs or other antiaggregants such as dipyridamole)17 might be necessary for primary thromboprophylaxis in aPL-positive patients. In this study, however, no association was found between APS manifestations and additional thrombotic risk factors.
One of the characteristic clinical manifestations of the APS is fetal morbidity, including early and late pregnancy losses, intrauterine growth restriction and prematurity. Additionally, maternal morbidity (pre-eclampsia, eclampsia and abruptio placentae) is also relatively common in pregnant patients with APS. Only a small percentage of patients from this cohort became pregnant during this study period (9.4%), compared with a higher proportion of patients with history of pregnancy at the study entry (71.9%).2 However, 81.8% of patients who became pregnant during the follow-up succeeded in having one or more live births and this is higher than the percentage of success in the previous period (74%).2
In 1992, Asherson18 described the existence of a new APS subset in which multiple vascular occlusive events, usually affecting small vessels supplying organs and presenting over a short period of time (less than a week), were the outstanding features. This subset was termed the “catastrophic” APS19 and it is considered one of the most severe complications of the APS, as about half of the patients succumb despite seemingly adequate treatment, including anticoagulation, steroids, plasma exchange and intravenous immunoglobulins.20,21 In this study, the incidence of this “catastrophic” subset in a 5-year period was found to be around 0.9% and the mortality rate 55.6% (5/9 patients).
In this study, neither individual aPL nor the combination of some of them was associated with an increased incidence of any specific clinical manifestation. Although previous studies2 have found occasional associations between aPL patterns and specific clinical manifestations, the lack of associations in this study may be due to the relatively small number of clinical manifestations during the study period.
The total mortality rate during this period was 5.3%, slightly higher than the mortality in a European SLE cohort of 1000 patients (“Euro-lupus cohort”) (4.5%) during a similar period.22 In addition to severe thrombotic events (ie, myocardial infarction, stroke or the catastrophic APS), infections and haemorrhages accounted for one-third of deaths. The finding of infections as a common cause of death in APS is of particular importance as these have been implicated in the aetiopathogenesis of the syndrome itself.23 However, it is important to emphasise that determining the main cause of death for patients with APS can be difficult, as many patients frequently present multisystemic involvement and infections appear as a final complication. In fact, the majority of infections (ie, pneumonias, sepsis) appeared in severely ill patients who had been admitted to intensive care units and were receiving multiple life-support treatment. We were not able to identify any clinical or immunological parameter with prognostic significance for mortality. Interestingly, older age was not associated with increased mortality and this may be owing to the fact the APS-related conditions (which are not age dependent) dominated the causes of mortality in this cohort, but also because the population was not very old.
In conclusion, our study provides updated information on APS morbidity and mortality characteristics in this decade. Patients with APS still develop significant morbidity and mortality despite current treatment (mainly, oral anticoagulants and/or antiaggregant agents). These findings call for an increased effort in determining optimal prognostic markers and therapeutic measures to prevent these important complications in the APS.
Appendix A: the Euro-Phospholipid Project Group (European Forum on Antiphospholipid Antibodies)
The members of the Euro-Phospholipid Project Group (European Forum on Antiphospholipid Antibodies) are as follows: Coordinators are Ricard Cervera, Josep Font, Jean-Charles Piette, Marie-Claire Boffa, Munther A Khamashta and Graham R V Hughes; from Hospital Clínic, Barcelona, Catalonia, Spain, Ricard Cervera, Gerard Espinosa, Silvia Bucciarelli, Manuel Ramos-Casals, Albert Bové, Angel Robles and Juan Miguel Antón; from Hôpital Pitié-Salpêtrière, Paris, France, Jean-Charles Piette, Camille Francès, Zahir Amoura and Marie-Claire Boffa; from St Thomas’ Hospital, London, UK, Munther A Khamashta, Cecilia N Pisoni, María Laura Bertolaccini and Graham RV Hughes; from Hospital Regional “Carlos Haya”, Málaga, Spain,María Teresa Camps and Enrique de Ramón; from Chaim-Sheba Medical Center, Tel-Hashomer, Israel, Yehuda Shoenfeld and Gisele Goddard; from Copenhagen University Hospital at Rigshospitalet, Copenhagen, Denmark, Soren Jacobsen; from Medical and Health Science Centre, Debrecen, Hungary, Gabriella Lakos, Emese Kiss, Pal Soltesz and Margit M Zeher; from Spedali Civili, Brescia, Italy, Angela Tincani and Marco Taglietti; from Hippocration Hospital, Athens, Greece, Irene Kontopoulou-Griva, G Theodossiades and Eufrosyni Nomikou; from Policlinico “Le Scotte”, Siena, Italy, Mauro Galeazzi and Francesca Bellisai; from Istituto Auxologico, Milan, Italy, Pier Luigi Meroni and Cristina Luzzana; from University Medical Center, Utrecht, The Netherlands, Ronald H W M Derksen and Philip G de Groot; from Immanuel-Krankenhaus GmbH, Berlin, Germany, Erika Gromnica-Ihle; from Medical University, Sofia, Bulgaria, Marta Baleva; from Università di Pisa, Pisa, Italy, Stefano Bombarderi and Marta Mosca; from Cliniques Universitaires Saint-Luc, Université Catholique de Louvain, Brussels, Belgium, Frédéric Houssiau and Chantal Lefèbvre; from Centre Hospitalier Universitaire, Nìmes, France, Jean-Christophe Gris and Isabelle Quéré; from Hôpital Claude Huriez, Lille, France, Eric Hachulla and Monique Tomczak; from Hospital Geral San António, Porto, Portugal, Carlos Vasconcelos, Paulo Barbosa, Isabel Almeida, Fatima Farinha and Manuel Campos; from Technische Universität Dresden, Dresden, Germany, Beate Roch; from Hospital Clínico Universitario, Málaga, Spain, Antonio Fernández-Nebro, Manuel de Haro and Manuel Abarca.
REFERENCES
Footnotes
For numbered affiliations see end of article
Josep Font (a member of the Euro-Phospholipid Project Group) died during the preparation of this manuscript and the authors want to dedicate this article to his memory.
A complete list of members of the Euro-Phospholipid Project Group (European Forum on Antiphospholipid Antibodies) is given in Appendix A.
Funding Supported in part by grants FISS 2003/028 from Fondo de Investigaciones Sanitarias of Spain, Ricerca Corrente 2000 from IRCCS Istituto Auxologico Italiano of Italy and from Institut Electricité-Santé of France.
Competing interests None.
Ethics approval Ethics committee approval obtained.