Article Text

Abstract
Objective: To assess the relationships between serum B lymphocyte stimulator (BLyS) levels, autoantibody profile and clinical response in patients with systemic lupus erythaematosus (SLE) following rituximab-based B cell depletion therapy (BCDT).
Methods: A total of 25 patients with active refractory SLE were followed for ⩾1 year following BCDT. Disease activity was assessed using the British Isles Lupus Assessment Group (BILAG) system, and serum levels of BLyS and autoantibodies to dsDNA and extractable nuclear antigens (ENA) measured by ELISA. Serum immunoglobulins and anti-dsDNA antibodies were assessed for expression of the 9G4 idiotope (indicating VH4–34 germline gene origin).
Results: Following BCDT, all patients depleted in the peripheral blood and improved clinically for ⩾3 months. Pre-BCDT BLyS levels were quantifiable (median 1.9 ng/ml) in 18/25 patients and rose in most patients at 3 months post-BCDT (median 4.15 ng/ml). Nine patients, all with quantifiable pre-BCDT serum BLyS, experienced a disease flare within 1 year. This group of patients was more likely to harbour anti-Ro/SSA antibodies (odds ratio 1.76; p = 0.06) with higher serum levels (p = 0.0027; Mann–Whitney U test). Serum levels of anti-ribonucleoprotein (RNP)/Sm were also higher in this group (p<0.05). Expression of VH4–34 by serum immunoglobulins and anti-dsDNA antibodies had no predictive value for the length of clinical response.
Conclusions: Patients with SLE with an expanded autoantibody profile and raised BLyS levels at baseline had shorter clinical responses to BCDT. This may reflect a greater propensity to, and degree of, epitope spreading in such patients and suggests that treatment regimens beyond BCDT may be necessary to induce long-lasting clinical remissions in these individuals.
Statistics from Altmetric.com
The availability of the B cell-targeting anti-CD20 monoclonal antibody (mAb) rituximab has enabled the development of B cell depletion therapy (BCDT) for the treatment of autoimmune rheumatic diseases. Open-label studies in systemic lupus erythaematosus (SLE) have shown promising results.1 In treated patients, clinical response often extends for months or even years beyond the period during which peripheral blood B cells are depleted.2 Given that re-population of the peripheral blood by B cells does not always herald disease flare, the qualitative properties of the re-populating B cell clones may be central to the reinstatement of pathogenic mechanisms. Thus, association of disease return or flare with B cell return may reflect the rapid re-expansion of pathogenic B cell clones. Alternatively, a delay between restoration of peripheral blood B cell numbers and clinical relapse or flare may indicate a more gradual accumulation of those B cell clones with pathogenic potential.
Accordingly, the mechanisms that permit or facilitate persistence of putative pathogenic B cell clones and/or plasma cells are of great relevance to optimising the clinical application of BCDT. Achieving adequate depletion of B cells presents a greater problem in patients in whom complement or cellular cytotoxicity (through Fcγ-receptors) mechanisms are compromised, as rituximab itself is relatively ineffectual at signalling cell death. Additional cytotoxic agents, in conjunction with rituximab, may be needed to induce effective depletion. Abnormal levels of the B cell survival factor (B lymphocyte stimulator (BLyS), also known as BAFF), have also been associated with human autoimmune diseases.3 Circulating levels of biologically active BLyS are elevated in some patients with rheumatoid arthritis (RA), SLE and Sjögren syndrome,4 and elevated circulating BLyS levels modestly correlate with disease activity in patients with SLE.5 In animal models, BLyS overproduction promotes development of autoimmunity in non-autoimmune-prone hosts6 and accelerates autoimmunity in autoimmune-prone hosts.7 Murine models have also shown that excessive BLyS levels may promote survival of self-reactive B cells by affecting selection stringency.8 Thus, elevated BLyS levels, by facilitating amplification and spreading of the autoimmune response, may promote re-emergence and/or re-expansion of pathogenic autoreactive B cells following BCDT, thereby leading to more rapid recurrence of disease. We have, therefore, examined the relationships among serum BLyS levels, autoantibody profiles and clinical responses in patients with SLE undergoing BCDT. The presence of a specific autoreactive B cell subpopulation, namely B cells utilising the immunoglobulin variable heavy chain gene VH4-34, recognised by the rat monoclonal antibody 9G4, was also determined.9
PATIENTS AND METHODS
Patients
The 25 patients with SLE, who all met ⩾4 of the revised criteria of the American College of Rheumatology (ACR), formed part of a larger cohort treated with BCDT. Clinical details and circulating antibody responses in some have previously been reported.10 11 Briefly, all patients had clinically active SLE with diverse clinical features, and had failed standard immunosuppressive therapy. All received two doses of intravenous rituximab (each dose 500 mg for patients 2, 3, 4 and 6; each dose 1000 mg for all other patients) 2 weeks apart. In all except three patients (numbers 9, 12 and 27; one refused, one had an allergy and one remained on methotrexate), rituximab was given in conjunction with two doses of intravenous cyclophosphamide (each dose 750 mg), and all patients received a variable dose of prednisolone (total dose of oral or intravenous steroid 200–500 mg within 3 weeks; average 250 mg per patient). Disease activity was evaluated using the British Isles Lupus Assessment Group (BILAG) index, which distinguishes activity in eight organs or systems.12 Activity in each system is graded from A (highest) to E (lowest). In this study, a flare was defined as development of a BILAG Grade A (major flare) or Grade B (moderate flare) in any system in which the previous assessment was C, D, or E. The categories were combined as both grades of flare required additional therapeutic intervention. Serum samples were collected and stored at −80°C and clinical evaluations were performed prior to BCDT, at least twice during the first 6 months after BCDT, and, where possible, every 2–3 months afterwards. In six patients, samples were also collected at 1 month post-BCDT. The patients had all given informed consent.
Patients were arbitrarily divided into those who suffered a disease flare within 1 year of BCDT and those who did not flare for more than 1 year. Patients were assigned to the “flare” group as soon as a flare had occurred. The cut-off point of 1 year was chosen to give a generous time period for a patient with lupus not to require any additional treatment in response to one cycle of BCDT. Patients were routinely encouraged (as always) to be seen by a doctor before a scheduled appointment if they had any concern about their disease, thereby minimising the chance of missing a flare.
Assessment of B cell depletion and B cell return
The normal range for CD19+ B cells used by the local pathology laboratory was 0.03−0.40×109/litre. Levels <0.005×109/litre were defined as undetectable. Depletion of B cells in the peripheral blood was deemed to have occurred when CD19+ B cells were undetectable. In all patients, total peripheral blood B cell depletion was achieved for at least 2 months. “B cell return” was defined as when B cells were again detectable in the peripheral blood (ie, when the CD19+ cell count was ⩾0.005×109/litre).
Serum levels of BlyS
Serum levels of BLyS were measured by ELISA6 using Fab fragments of the capture antibody rather than the whole antibody to reduce assay interference from rheumatoid factor. Samples with BLyS concentrations below the lower limit of quantification (BLOQ; 0.858 ng/ml) were arbitrarily assigned a value of 0.75 ng/ml for statistical and graphic purposes.
Serum levels of autoantibodies
ELISAs were used to detect antibodies to dsDNA (phage), histones, Ro/SSA, Sm and ribonucleoprotein (RNP)/Sm using the EL-ANA profiles commercial system (Theratest labs Inc. Lombard, Illinois, USA) and VH4–34 usage by anti-dsDNA antibodies measured as previously described.13 Expression of VH4–34 on circulating total IgM and total IgG was measured using a capture ELISA technique. Briefly, one side each of two ELISA plates were coated with goat anti-human IgM (1 μg/ml) or goat Fab2 anti-hIgG (0.5 μg/ml). After addition of serum samples to coated and uncoated sides of each plate, sequential additions of 9G4 reagent (10 μg/ml), goat anti-(rat–biotin) conjugate and streptavidin–horseradish peroxidase and colour development with tetramethylbenzidine followed. Mean + 3SD of the optical density (OD) given by eight normal control sera were calculated as the cut-off for normal values (table 1).
Statistical analysis
All calculations were performed using GraphPad Prism software for Macintosh (GraphPad, San Diego, California, USA). Differences in levels between the two groups (those who did not flare for at least 1 year post-BCDT and those with a flare during the first year) with respect to the parameters of interest were assessed using the Mann–Whitney U test and Fisher exact test. The Wilcoxon signed rank (paired samples) test was used to compare BLyS levels pre- and post-BCDT. Baseline immunological results of patients in each group were analysed using logistic regression to identify any possible independent or multiple predictors of flare.
RESULTS
Immunological parameters at baseline
Table 1 summarises the organ involvement and serology of the 25 patients with SLE prior to BCDT (patient numbers correspond with those published previously).11 Patients were grouped into those with no flare for at least 1 year post-BCDT (n = 16) and those with an “early” flare during the first year post-BCDT (n = 9). Quantifiable pre-BCDT serum BLyS levels were significantly associated with early flare (9/9 with early flare vs 9/16 without flare for >1 year; Fisher exact test p = 0.027). The median pre-BCDT serum BLyS level was higher in those patients who experienced an early flare (median: 3.75 ng/ml) than in those that did not (median: 1.46 mg/ml), but this did not achieve statistical significance. Logistic regression revealed an association between the presence of serum anti-Ro/SSA autoantibodies and a shorter clinical response (odds ratio: 1.76; p = 0.06), but not with the presence of any of the other tested autoantibodies including antibodies to Sm or RNP/Sm (odds ratios of 0.56 and 0.69, respectively). No significant correlations were detected between BLyS levels and individual extractable nuclear antigen (ENA) specificities (data not shown). In addition, as all patients who flared within the first year had detectable BLyS, this confounded analyses of variance between BLyS and associated factors. It was however notable that in the 9 patients with a flare <1 year, 7 of these had raised BLyS as well as being positive for anti-Ro antibodies whereas in patients with a remission greater than 1 year, only 4 of 16 possessed both at baseline (p = 0.017, Fisher exact test). When pre-treatment levels of the different autoantibodies were compared between the two groups of patients (fig 2A–F), levels of anti-Ro/SSA and anti-Sm were significantly higher in those patients with an early flare (p = 0.0027 and p = 0.041, respectively, Mann–Whitney U rank sum) along with a tendency for antibodies to RNP/Sm to also be higher (p = 0.075). In contrast, serum levels of anti-dsDNA or anti-histone antibodies did not segregate with development of disease flare within 1 year of BCDT.

Effect of BCDT on circulating BLyS levels
At baseline, 18/25 patients had quantifiable circulating BLyS levels (table 1). In the patient group as a whole, serum BLyS levels had significantly increased from a pre-BCDT median of 1.9 ng/ml to a 3-month post-BCDT median of 4.15 ng/ml (p = 0.0028, fig 1A). By 6–8 months post-treatment, most of the patients had repopulated their B cells in the peripheral blood, and the median serum BLyS levels had decreased to 1.65 ng/ml (p<0.001 compared to 3 months).
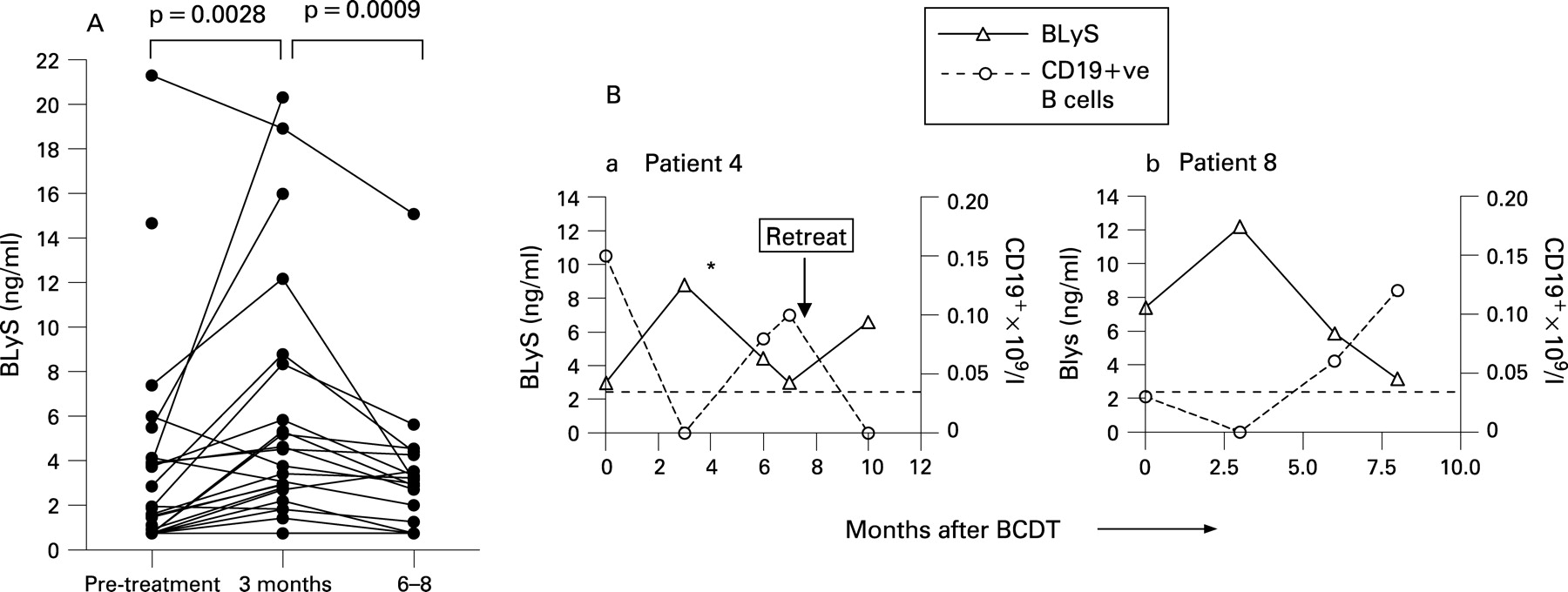
Relationship between B cell repopulation and circulating BLyS levels
Similar to our observations in patients with RA treated with BCDT (13), the fall in serum BLyS levels by 6–8 months post-BCDT was associated with return of B cells to the peripheral blood, which occurred between 2 and 10 months after BCDT in 19 of 21 patients from whom data were available (table 1). In general, B cell counts were within the normal range when repopulating B cells were first detected, and serum BLyS levels at B cell repopulation were similar in patients in whom B cell return was accompanied by disease flare and those who remained well (data not shown). There was no relationship between BLyS levels and length of time to repopulation (p = 0.81, r2 = 0.003). Representative experiments showing serial peripheral blood and CD19+ cell counts and serum BLyS levels are shown for two patients (fig 1B). Serum BLyS levels and B cell counts followed antiparallel courses in each patient.
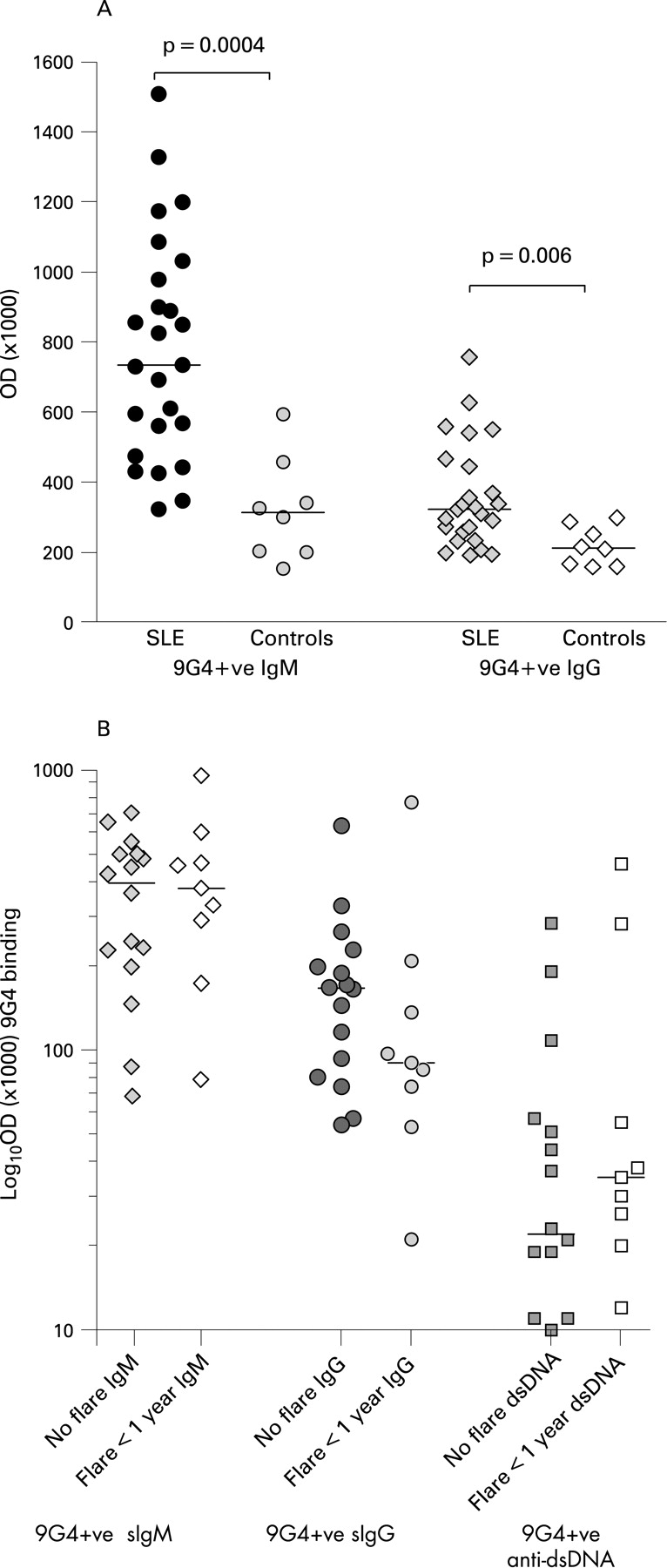
No association of VH4–34 usage with early relapse
As previously described,9 9G4 expression was confirmed to be significantly higher on serum IgM and IgG in patients with SLE compared with normal controls (p = 0.004 and p = 0.006, respectively; Mann–Whitney U rank sum test) (fig 3A). There was, however, no difference in the level of 9G4 expression on IgM, IgG or anti-dsDNA antibodies between those patients with a disease flare within 1 year of BCDT or those with a more lengthy response to BCDT (fig 3B and table 1).

{kind=link}
{kind=link}
{kind=link}
DISCUSSION
In order to maximise the efficacy of B cell-targeted therapies in patients with SLE, it is important to identify key biomarkers that contribute to the disease process and/or that identify those patients most likely to benefit from this form of therapy. We have previously shown that a good clinical response is associated with a decrease in circulating anti-dsDNA antibodies, but not anti-ENA or antimicrobial antibodies, in patients with SLE who achieved adequate peripheral blood B cell depletion following BCDT.14 This suggests that some autoantibody-producing B cell clones are protected from rituximab-mediated cytotoxicity, perhaps due to difficulties in vascular access or the presence of local pro-survival signals.15 Alternatively, most of the autoantibody production for certain specificities may be due to mature, fully differentiated plasma cells that do not express CD20 and, hence, are resistant to CD20-directed therapy. We have previously shown that anti-dsDNA antibodies expressing the 9G4 idiotope may be more sensitive to BCDT.14 The more rapid disappearance of 9G4+ anti-dsDNA antibodies after depletion may reflect their production mainly by short-lived plasma cells. The present study did not however show whether 9G4 expression on serum total IgM, serum total IgG, or anti-dsDNA antibodies were useful predictors of clinical response (flare within 1 year or not).
In this observational study of 25 patients with SLE, there was a strong association between a more protracted response to BCDT in those patients with raised baseline serum levels of BLyS. This was probably not simply the result of more aggressive treatment of those patients with the shorter responses to BCDT. Cytotoxic drugs do not appear to influence BLyS levels and high-dose glucocorticoids may actually cause a decline in BLyS levels.16 We also did not observe an inverse relationship between BLyS levels and repopulation as described in patients with Sjögren syndrome.17 An association was found between both the presence of, and relatively higher levels of, autoantibodies to Ro/SSA and to Sm, with a shorter clinical response. This may indicate epitope spreading to a wider number of antigenic specificities, perhaps under the influence of raised BLyS levels. The lack of association of baseline levels of anti-dsDNA antibodies with clinical response may relate to the fact that anti-dsDNA-committed B cells behave differently from anti-ENA committed B cells. As previously described, anti-dsDNA antibodies generally decline following BCDT whilst those directed against ENA do not respond as readily.14 This suggests that at least a proportion of anti-dsDNA antibodies are derived from a B cell subpopulation that only gives rise to short-lived (possibly extra-follicular) plasma cells.
Studies in mice have strongly suggested that limited circulating levels of BLyS may favour survival of non-autoreactive B cells over survival of autoreactive B cells.8 The apparent association of the presence of antibodies to Ro/SSA and possibly also other ENA specificities with a shorter clinical response to BCDT raises the question of whether as well as being predictive diagnostic and disease severity markers, some ENA-specific B cell clones or autoantibodies may play a more direct role in disease progression or persistence. Internalisation of nucleic acids and associated proteins by BCR expressed on specific memory B cells allows access to endosomal Toll-like receptors (TLR; DNA is focussed to TLR-9 and TLR-7 binds ssRNA). With IgG autoantibodies resulting from this interaction, it is possible that immune complexes containing DNA or RNA are able to be taken up through Fc receptors (notably FcγRIIa) on antigen presenting cells, in particular plasmacytoid dendritic cells (pDC).18 The triggering of TLR signalling within pDC results in the production of large quantities of type 1 interferon and interleukin (IL)6.19 This, in turn, contributes to B cell activation and maturation and completes a vicious cycle. We have previously shown that ENA levels are relatively resistant to BCDT, particularly in comparison with anti-dsDNA antibodies.14 Here we have shown that the possession of an expanded repertoire of ENA, in particular those of Ro/SSA specificity, are associated with a shorter clinical response to BCDT. This suggests that plasma cells committed to ENA autoantigens are long-lived, perhaps as a result of the elicitation of more robust T cell help for RNA-associated proteins. The continued presence of autoantibodies (against ENA), despite peripheral B cell depletion, may therefore allow immune complex formation (if sufficient antigen is available) and allow more rapid re-institution of a vicious cycle in these patients.
As we have previously found in patients with RA following BCDT, serum BLyS levels rose rapidly whether or not pre-treatment levels were elevated, and these levels remained raised for the period of B cell depletion or longer.13 This rise was thought to reflect the reduction of B cell load but may also include the contribution of a homeostatic mechanism. Many of the abnormalities of B cell function that are well recognised features of human SLE have now been shown to be regulated or affected by BLyS. Although this does not prove that dysregulation of BLyS underlies the immunopathogenic characteristics of SLE, it does suggest possible new paths for intervention. The increased prevalence of raised baseline BLyS levels among those individuals who flared within the first year post-rituximab suggests that increased BLyS levels may help identify those patients who are at greatest risk for flare within the first year post-BCDT.
Additional studies in larger cohorts of patients will be needed to confirm whether serum BLyS and/or anti-ENA levels can be used as predictive parameters with which to profile patients with SLE for their duration of response to BCDT.
REFERENCES
Footnotes
Competing interests: MJL has received funding from GlaxoSmithKline Research and Development Ltd and from Roche Pharmaceuticals. MT is a major stockholder and a paid executive of TheraTest Labs Inc. JCWE has received financial support from Roche Pharmaceuticals and Genentech. WS has received consulting fees from Human Genome Sciences. TSM was an employee of Human Genome Sciences while conducting this study. DAI and GC have no competing interests.