Article Text

Abstract
Objective: To examine the relative importance of tumour necrosis factor-receptor 1 (TNF-R1) and TNF-R2 and their signalling pathways for pro-inflammatory and pro-destructive features of early-passage synovial fibroblasts (SFB) from rheumatoid arthritis (RA) and osteoarthritis (OA).
Methods: Cells were stimulated with tumour necrosis factor (TNF)α or agonistic anti-TNF-R1/TNF-R2 monoclonal antibodies. Phosphorylation of p38, ERK and JNK kinases was assessed by western blot; proliferation by bromodesoxyuridine incorporation; interleukin (IL)6, IL8, prostaglandin E2 (PGE2) and matrix metalloproteinase (MMP)-1 secretion by ELISA; and MMP-3 secretion by western blot. Functional assays were performed with or without inhibition of p38 (SB203580), ERK (U0126) or JNK (SP600125).
Results: In RA- and OA-SFB, TNFα-induced phosphorylation of p38, ERK or JNK was exclusively mediated by TNF-R1. Reduction of proliferation and induction of IL6, IL8 and MMP-1 were solely mediated by TNF-R1, whereas PGE2 and MMP-3 secretion was mediated by both TNF-Rs. In general, inhibition of ERK or JNK did not significantly alter the TNFα influence on these effector molecules. In contrast, inhibition of p38 reversed TNFα effects on proliferation and IL6/PGE2 secretion (but not on IL8 and MMP-3 secretion). The above effects were comparable in RA- and OA-SFB, except that TNFα-induced MMP-1 secretion was reversed by p38 inhibition only in OA-SFB.
Conclusion: In early-passage RA/OA-SFB, activation of MAPK cascades and pro-inflammatory/pro-destructive features by TNFα is predominantly mediated by TNF-R1 and, for proliferation and IL6/PGE2 secretion, exclusively regulated by p38. Strikingly, RA-SFB are insensitive to p38 inhibition of MMP-1 secretion. This indicates a resistance of RA-SFB to the inhibition of pro-destructive functions and suggests underlying structural/functional alterations of the p38 pathway, which may contribute to the pathogenesis or therapeutic sensitivity of RA, or both.
- ARA, American Rheumatism Association
- BrdU, bromodesoxyuridine
- FCS, fetal calf serum
- IL, interleukin
- DMEM, Dulbecco’s modified Eagle’s medium
- mAbs, monoclonal antibodies
- MAPK, mitogen-activated protein kinases
- MMP, matrix metalloproteinase
- OA, osteoarthritis
- PBS, phosphate-buffered saline
- PGE2, prostaglandin E2
- RA, rheumatoid arthritis
- SFB, synovial fibroblasts
- TNF, tumour necrosis factor
- TNF-receptor
- synovial fibroblast
- p38 MAP kinase
- interleukin
- matrix metalloproteinase
Statistics from Altmetric.com
- ARA, American Rheumatism Association
- BrdU, bromodesoxyuridine
- FCS, fetal calf serum
- IL, interleukin
- DMEM, Dulbecco’s modified Eagle’s medium
- mAbs, monoclonal antibodies
- MAPK, mitogen-activated protein kinases
- MMP, matrix metalloproteinase
- OA, osteoarthritis
- PBS, phosphate-buffered saline
- PGE2, prostaglandin E2
- RA, rheumatoid arthritis
- SFB, synovial fibroblasts
- TNF, tumour necrosis factor
In rheumatoid arthritis (RA), activated synovial fibroblasts (SFB) contribute to the inflammatory/destructive potential of the aggressive pannus tissue by producing pro-inflammatory mediators and matrix-degrading enzymes.1–6
Tumour necrosis factor α (TNFα), a pro-inflammatory cytokine with a critical role in RA, is primarily produced by monocytes/macrophages and expressed as a bioactive 26 kDa precursor transmembrane molecule or a secreted mature 17 kDa cytokine.7,8 The biological activity of TNFα is mediated by binding to two distinct but related receptors of 55–60 kDa (TNF-R1) and 75–80 kDa (TNF-R2).7,9 TNFα binding to its receptors induces the activation of several signal transduction cascades.7 Besides the NF-κB pathway, the mitogen-activated protein kinases (MAPK) play an essential role for the TNFα signalling. This signalling cascade contains three pathways: the p38 MAP kinase (p38), Erk kinase (ERK), and Jun kinase (JNK) pathway. They are activated by serine/threonine and tyrosine phosphorylation. The important role of these signal transduction pathways has been shown in several animal models.10–12 In human SFB, activation of all three signalling pathways by TNFα has been reported.13 Inhibition of the p38 and JNK pathway in RA-SFB decreased the expression and synthesis of TNFα-induced pro-destructive/pro-inflammatory molecules.13,14 Therefore, these signalling cascades are regarded as highly attractive targets for new anti-inflammatory drugs in RA.10 Differential effects of the TNF-Rs on the pro-destructive and pro-inflammatory character of cells have been previously studied. In skin fibroblasts, the stimulation of TNF-R1 induces expression of matrix metalloproteinase (MMP)-1 and MMP-3.15 In RA-SFB, TNF-R1 stimulation prompts secretion of interleukin (IL)6, IL8, prostaglandin E2 (PGE2) and MMP-1.16,17
Expression of both TNF-Rs has been shown in RA synovial tissue18 and on RA- and osteoarthritis (OA)-SFB.16,17,19,20 Although the central role of MAPK pathways in the TNFα-induced synthesis of pro-inflammatory/pro-destructive properties has been described,14,21 their specific importance for signal transduction through the two different TNF-Rs in SFB and, in particular, early-passage SFB, has not been studied to date.
In this study, early-passage RA-SFB were therefore compared with OA-SFB concerning the relative importance of TNF-R1/TNF-R2 for TNFα-induced signalling of the MAPK pathways, proliferation, secretion of IL6, IL8, prostaglandin E2 (PGE2) and matrix metalloproteinase-1 (MMP-1) as well as the sensitivity of these functions to inhibition of p38, ERK and JNK. The effects were assessed using agonistic anti-TNF-R antibodies. Early passage SFB were used for this study to minimise the influence of repeated passages on the expression/secretion of effector molecules and to avoid the accumulation of chromosomal aberrations which may also influence gene expression.22,23
PATIENTS AND METHODS
Patients
Synovial tissue from patients with RA and OA was obtained during open joint replacement/arthroscopic synovectomy from the Clinic of Orthopaedics, Eisenberg, Germany. All patients fulfilled the respective American Rheumatism Association (ARA) criteria.24,25 Informed consent was obtained from patients and the study was approved by the ethics committee of the University of Jena, Germany. In total, synovial tissues from 36 patients (18 RA and 18 OA) were used in this study. All OA synovial tissues were obtained from knee joints. The RA synovial tissues were also obtained from knee joints, except for two samples, one each from the ankle and the wrist joint. Immediately after synovectomy, tissue was placed in culture medium at ambient temperature and subjected to digestion within 2 hours.
Tissue digestion and cell culture
RA and OA synovial samples were digested and subsequently cultured for 7 days, as previously described.22,26 SFB were negatively isolated from trypsinised RA and OA primary-culture synovial cells as previously published.22 SFB were cultured in the virtual absence of contaminating non-adherent cells and macrophages. Mycoplasma contamination of the cells was excluded by 4′-6-diamidino-2-phenylindole staining.
For stimulation experiments, cells were seeded at the indicated concentrations and cultured for 24 hours in Dulbecco’s modified Eagle’s medium (DMEM)/10% fetal calf serum (FCS) at 37°C and 5% CO2. Thereafter, cells were incubated with TNFα (10 ng/ml), agonistic anti-TNF-R1 (HTR-9; 10 μg/ml), or agonistic anti-TNF-R2 monoclonal antibodies (mAbs; UTR-1; 10 μg/ml) in DMEM/0.2% lactalbumin hydrolysate for the indicated times.27,28 For inhibition of the individual MAPK pathways, cells were preincubated for 30 minutes with SP203580 (1 μmol/l), U0126 (1 μmol/l; both from Alexis, Grünberg, Germany), or SP600125 (20 μmol/l; Calbiochem, Schwalbach, Germany). In all cases, phosphorylation of the respective MAPK was consistently reduced by the inhibitors, indicating effective inhibition.
Analysis of TNF-R expression by flow cytometry
Second passage RA- and OA-SFB were analysed by flow cytometry for surface expression of TNF-R1 and TNF-R2 using the anti-TNF-R1 mAb H398 and the anti-TNF-R2 mAb 80M2 (10 μg/ml each). SFB were seeded at a concentration of 2.5×105 cells/well in 24-well plates and allowed to adhere for 24 hours. Thereafter, cells were trypsinised and analysed for TNF-R1/R2 surface expression, as previously described.17,22 In addition, cells were cultivated for 24 hours with/without 10 ng/ml TNFα (R&D Systems, Wiesbaden, Germany), followed by trypsinisation of the cells and FACS analysis.
Analysis of the TNFα/TNF-R-induced signal transduction by western blotting
For kinetic analysis of MAPK activation, RA-SF (beginning of third passage; 1×106 cells/well of a 12-well plate, derived from three patients) was stimulated with TNFα for 0, 5, 10, 20 and 40 minutes. At the end of the incubation time, cells were washed twice with ice-cold phosphate-buffered saline (PBS) and subsequently lysed with buffer for protein extraction (50 mM Tris, 150 mM NaCl, EDTA, pH 7.4, containing 100 mM NP40, 1 mM phenylmethylsulphonylfluoride, 1 mM Na3VO4, 2 μg/ml aprotinin, 2 μg/ml pepstatin and 2 μg/ml leupeptin). Samples were kept at −80°C until the analyses were performed.
For analysis of signal transduction, RA- or OA-SFB (beginning of third passage; 1×106 cells/well of a 12-well plate, derived from four patients each) were stimulated with TNFα or agonistic anti-TNF-R1/2 mAbs as described above for 10 or 20 minutes. At the end of the incubation time, cells were washed twice with ice-cold PBS and subsequently lysed with buffer for protein extraction. Samples were kept at −80°C until the analyses were performed.
A total of 25 μg of protein extract was separated by denaturing sodium dodecyl sulphate-polyacrylamide gel electrophoresis and transferred onto nylon membranes. After blocking with 2.5% skim milk in Tris-buffered-saline-Tween, membranes were probed with specific primary antibodies against the phosphorylated form of p38, ERK and JNK (BD Transduction Laboratories, Heidelberg, Germany, or Cell Signaling, Danvers, MA, USA) overnight at 4°C, washed, incubated with horseradish peroxidase-conjugated IgG as secondary antibody, and visualised by chemiluminescence (Supersignal; West Chemiluminescent Substrate; Pierce, Rockford, USA). To determine even transfer and equal loading, membranes were stripped and reprobed with antibodies specific for the non-phosphorylated forms of p38, ERK and JNK. The intensity of each band was quantified using integration image software (Scion Corporation, Frederick, MD, USA).
Assessment of proliferation (bromodesoxyuridine (BrdU) assay)
Proliferation was assessed by BrdU incorporation using a commercially available cell proliferation ELISA (Roche, Mannheim, Germany). RA- or OA-SFB (n = 10 each; second passage, 3×103 cells/well of a 96-well microtitre plate) were stimulated as described above. After 24 hours’ incubation, BrdU was added (final concentration 10 μmol/l BrdU), followed by an additional incubation step for 24 hours at 37°C in 5% CO2. The incorporation of BrdU was detected according to the manufacturer’s instructions. The resulting colour was read at 450 nm in a microtitre plate spectrophotometer (Fluostar Optima; BMG Labtechnologies, Offenburg, Germany).
Analysis of the TNFα/TNF-R-induced protein secretion of pro-destructive/pro-inflammatory molecules
RA- or OA-SFB (beginning of second passage; 2.5×105 cells/well of a 24-well plate, derived from six patients each for determination of MMP-1, IL6, IL8 and PGE2) were stimulated as described above. Thereafter, supernatants were collected. Samples were kept at −80°C until the analyses were performed.
Human IL6, IL8, MMP-1 and PGE2 were measured in diluted cell culture supernatants using commercially available ELISA kits (OptEIA, BD Biosciences, Heidelberg, Germany; Biotrak, Amersham-Pharmacia Biotech Inc, Freiburg, Germany). The resulting colour was read at 450 nm in microtitre plates (Fluostar Optima).
MMP-3 was analysed in the supernatants of stimulated cells by western blot using an MMP-3-specific antibody (clone 50647; R&D Systems). Quantification was performed as described above.
Statistical analysis
The non-parametric Mann–Whitney test was applied to analyse differences between controls and individual stimuli, or between RA and OA. Significant differences were accepted for p⩽0.05. The software SPSS 12.0 (SPSS Inc; Chicago, IL, USA) was used.
RESULTS
Expression of TNF-R1 and TNF-R2 on RA- and OA-SFB
SFB expressed both TNF-R1 (RA-SFB: 38.8 (6.4)% positive cells; OA-SFB: 47.3 (8.2)%; fig 1A) and TNF-R2 (RA-SFB: 11.8 (3.3)%; OA-SFB: 7.8 (2.5)%; fig 1B). Stimulation of the cells for 24 hours with TNFα significantly reduced TNF-R1 expression on RA- and OA-SFB compared with non-stimulated cells cultured for 24 hours (RA-SFB: from 26.0 (5.5)% to 1.5 (0.6)%; OA-SFB: from 30.5 (4.8)% to 3.9 (1.7)%; both p⩽0.05). This was possibly due to TNF-R shedding or internalisation.29,30 In contrast, TNF-R2 expression on the cells was not significantly influenced by TNFα stimulation.

Analysis of TNF-R expression on the surface of RA- and OA-SFB after stimulation with TNFα. RA- and OA-SFB expressed TNF-R1 and TNF-R2 on their surface (A and B, one representative patient is shown). Only the TNF-R1 expression on the cells was significantly downregulated by stimulation with TNFα. *p<0.05 for the comparison of non-stimulated (0 hours) and TNFα-stimulated cells (24 hours); †p<0.05 for the comparison of non-stimulated (24 hours) and TNFα-stimulated cells (24 hours).
There were no significant differences between the percentages of TNF-R1- or TNF-R2-positive RA- and OA-SFB (whether non-stimulated or TNFα-stimulated), excluding that differential secretion of pro-destructive or pro-inflammatory molecules was due to differences in receptor expression.
Analysis of the TNFα/TNF-R-induced signal transduction in RA- and OA-SFB
Kinetics of MAPK activation
All three MAPK underwent maximal activation within 20 minutes after stimulation of RA-SFB (n = 3) with 10 ng/ml TNFα (fig 2). However, the kinetics of activation for the individual MAPK were different—that is, p38 was activated as early as 5 minutes after stimulation (maximum at 10 minutes; fig 2A), ERK activation was not induced until 10 minutes after stimulation (maximum at 20 minutes; fig 2B), and JNK was already activated 5 minutes after stimulation (maximum at 20 minutes; fig 2C). On the basis of these results, the time points 10 and 20 minutes were chosen for subsequent stimulations with TNFα/TNF-R mAbs; in the case of TNFα and TNF-R1 stimulation (see below), the results nicely reflected the kinetics shown in fig 2.
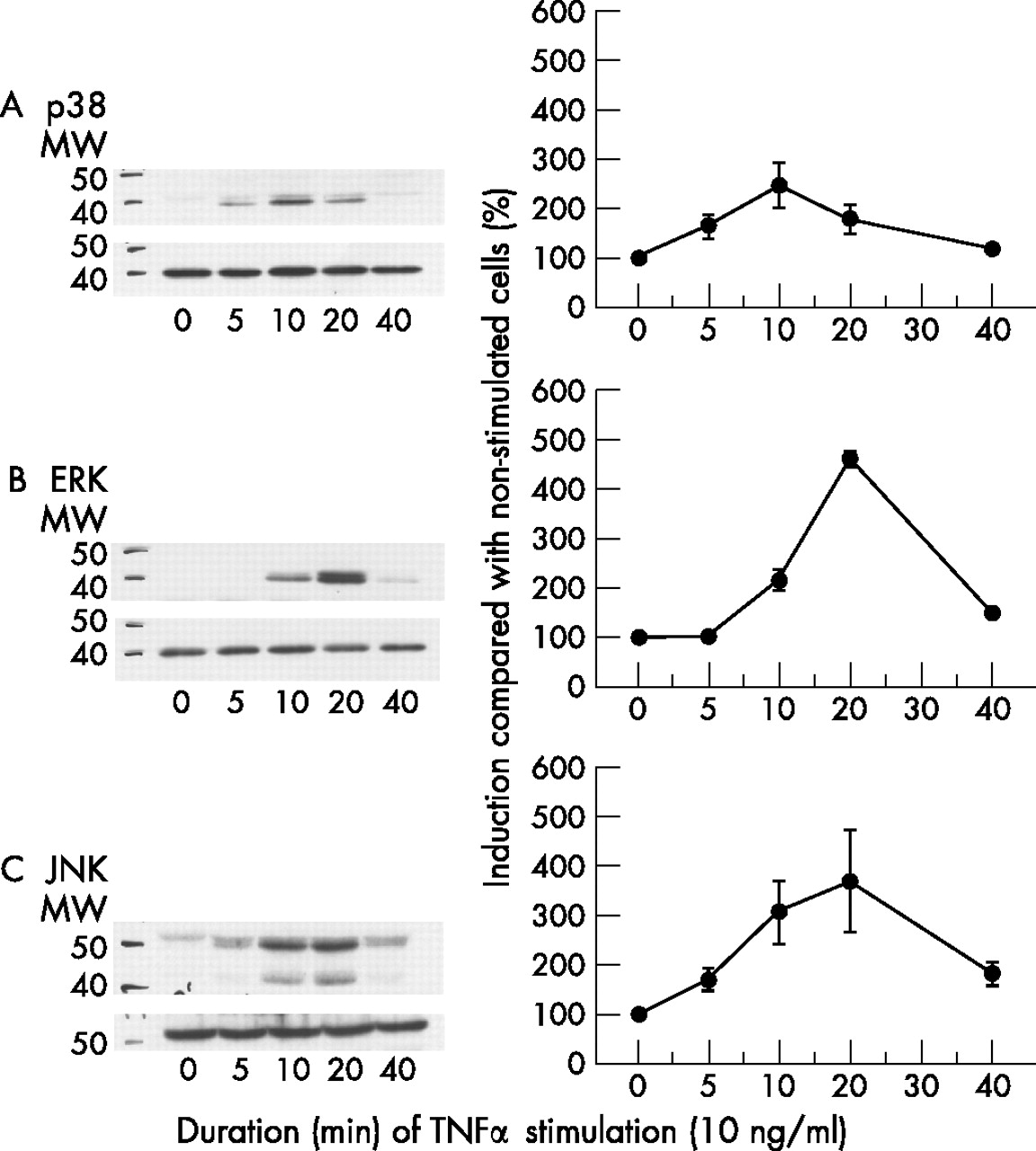
Time-dependent phosphorylation of p38 (A), ERK (B) or JNK (C) in third passage RA-SFB after stimulation with TNFα. All three MAPK underwent maximal activation within 10–20 minutes after stimulation of RA-SFB (n = 3) with 10 ng/ml TNFα. MW, molecular weight in kilodalton.
Activation of p38
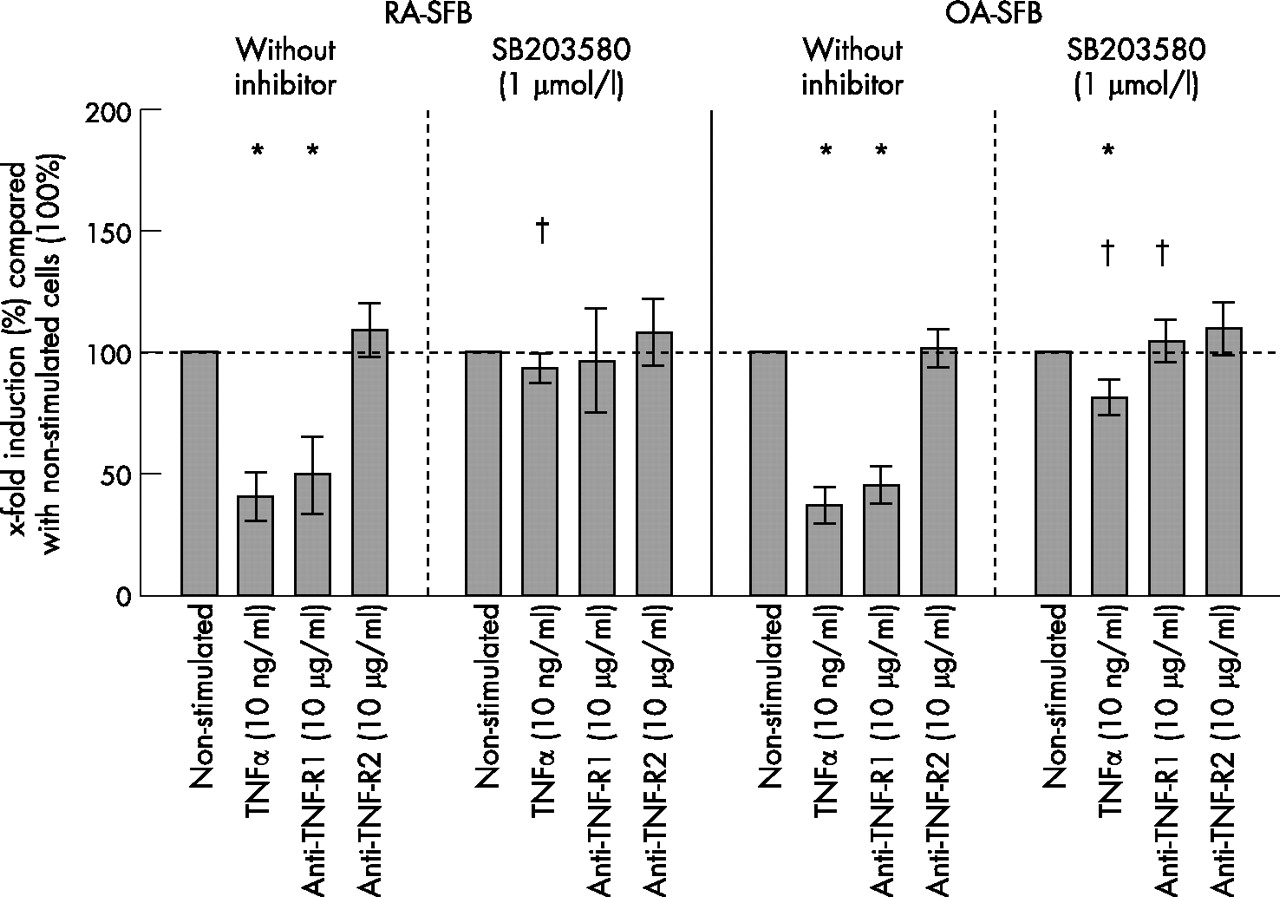
Stimulation of RA- and OA-SFB with TNFα for 10 or 20 minutes significantly induced the phosphorylation of p38 kinase compared with non-stimulated cells, without significant differences between RA and OA (RA-SFB: for 10 minutes’ stimulation 2.3-fold and for 20 minutes’ stimulation 2.9-fold induction; OA-SFB: for 10 minutes’ stimulation 2.1-fold and for 20 minutes’ stimulation 2.3-fold induction; shown for 20 minutes in fig 3A). This effect was exclusively mediated by TNF-R1 (ie, after the use of the agonistic anti-TNF-R1 mAb HTR-9).

Phosphorylation of MAP kinases in third passage SFB from patients with RA and OA after stimulation with TNFα, the agonistic anti-TNF-R1 mAb HTR-9, or the agonistic anti-TNF-R2 mAb UTR-1. Stimulation of RA- and OA-SFB (n = 4 each for p38 and JNK; n = 5 each for ERK) with TNFα for 20 minutes significantly induced the phosphorylation of p38 (A), ERK (B) and JNK kinase (C) through TNF-R1. Bars indicate means (SEM). *p<0.05 for the comparison of non-stimulated and stimulated cells; †p<0.05 for the comparison of RA- and OA-SFB.
Activation of ERK
Stimulation of RA- and OA-SFB with TNFα for 10 or 20 minutes significantly induced the phosphorylation of ERK compared with non-stimulated cells (RA-SFB: for 10 minutes’ stimulation 1.4-fold and for 20 minutes’ stimulation 4.0-fold induction; OA-SFB: for 10 minutes’ stimulation 1.2-fold and for 20 minutes’ stimulation 3.0-fold induction; shown for 20 minutes in fig 3B). This effect was exclusively mediated by TNF-R1. After 10 minutes’ stimulation, no significant group differences were observed. However, after 20 minutes’ stimulation with the agonistic anti-TNF-R1 mAb HTR-9, the phosphorylation was significantly higher in RA-SFB than in OA-SFB.
Activation of JNK
Stimulation of RA- and OA-SFB with TNFα for 10 minutes significantly induced the phosphorylation of JNK compared with non-stimulated cells only in RA-SFB but not in OA-SFB (RA-SFB: 3.4-fold; OA-SFB: 1.5-fold; data not shown). After 20 minutes’ stimulation, JNK phosphorylation was seen in both RA- and OA-SFB, without significant group differences (RA-SFB: 2.1-fold, OA-SFB: 2.4-fold; fig 3C). These effects were exclusively mediated by TNF-R1.
Influence of p38 MAP kinase, ERK kinase, or JNK kinase inhibition on TNF/TNF-R1-induced proliferation in RA- and OA-SFB
After stimulation with TNFα, RA- and OA-SFB showed a significantly reduced proliferation compared with non-stimulated cells (RA-SFB: 57% reduction of the mean; OA-SFB: 64% reduction; non-stimulated cells: 100%; fig 4), with no significant differences between RA- and OA-SFB. This effect was exclusively mediated by TNF-R1 (ie, after the use of the agonistic anti-TNF-R1 mAb HTR-9). In both RA- and OA-SFB, the reduction of proliferation by TNFα or selective TNF-R1 stimulation was almost completely abolished by inhibition of p38 (fig 4). In contrast, inhibition of ERK kinase by U0126 and JNK by SP600125 had no significant influence on the TNFα-induced reduction of proliferation in RA- and OA-SFB (data not shown). In all cases, no significant differences were seen between RA and OA.
Proliferation in second passage SFB from patients with RA and OA after stimulation with TNFα, the agonistic anti-TNF-R1 mAb HTR-9, or the agonistic anti-TNF-R2 mAb UTR-1 (with or without inhibition of p38). In RA- and OA-SFB (n = 6 each), TNFα stimulation significantly downregulated the proliferation compared with non-stimulated cells (100%). This effect was exclusively mediated by TNF-R1. This reduction was almost completely abolished by inhibition of the p38 MAPK with SB203580. Bars indicate means (SEM). *p<0.05 for the comparison of non-stimulated and stimulated cells; †p<0.05 for the comparison of cultures with or without SB203580.
Effects of selective TNF-R1/R2 stimulation on pro-destructive/pro-inflammatory features of RA- and OA-SFB
IL6 protein
RA- and OA-SFB (n = 6 each) constitutively secreted considerable amounts of IL6 (table 1; fig 5A). Stimulation with TNFα significantly induced IL6 secretion in both RA-SFB (55-fold) and OA-SFB (82-fold). This was exclusively mediated by TNF-R1, as shown by similar induction with the agonistic anti-TNF-R1 mAb HTR-9. Inhibition of p38 significantly reduced TNFα-augmented IL6 protein secretion in both RA- and OA-SFB (by about 42.3% and 56.3%, respectively). This effect was also observed after stimulation with the agonistic anti-TNF-R1 mAb HTR-9. Inhibition of ERK kinase by U0126 or JNK kinase by SP600125 did not significantly influence TNFα-induced IL6 secretion in either RA- or OA-SFB (data not shown). In all cases, no significant differences were seen between RA and OA.
Secretion of IL-6, IL-8, PGE2, and MMP-1 in RA- and OA-SFB following stimulation with TNF-α or with agonistic anti TNF-R1 (HTR-9) or anti TNF-R2 (UTR-1) mAbs with/without inhibition of the p38 MAP kinase with SB203580

IL6, IL8, and PGE2 secretion in second passage SFB from patients with RA and OA after stimulation with TNFα, the agonistic anti-TNF-R1 mAb HTR-9, or the agonistic anti-TNF-R2 mAb UTR-1 (with or without inhibition of p38). In RA- and OA-SFB (n = 6 each), TNFα significantly induced the secretion of IL6 (A), IL8 (B) and PGE2 (C) compared with non-stimulated cells through TNF-R1. The induction of IL6 (mediated through TNF-R1) and of PGE2 (mediated through both TNF-Rs) was significantly reduced by p38 inhibition in RA- and OA-SFB (A, C). Bars indicate means (SEM). *p<0.05 for the comparison of non-stimulated and stimulated cells; †p<0.05 for the comparison of cultures with or without SB203580.
IL8 protein
RA- and OA-SFB (n = 6 each) constitutively secreted considerable amounts of IL8 (table 1; fig 5B). Stimulation with TNFα significantly induced IL8 secretion in both RA-SFB (117-fold) and OA-SFB (95-fold). This was exclusively mediated by TNF-R1. Inhibition of p38 (SB203580), ERK (U0126) or JNK (SP600125) did not significantly influence TNFα-induced IL8 secretion (fig 5B; data not shown). In all cases, no significant differences were seen between RA and OA.
PGE2 secretion
RA- and OA-SFB (n = 6 each) secreted small amounts of PGE2 (table 1; fig 5C). Stimulation with TNFα significantly induced PGE2 secretion in both RA-SFB (66-fold) and OA-SFB (99-fold), an effect mediated by both TNF receptors with a predominance of TNF-R1 (ie, significant induction by the agonistic anti-TNF-R1 mAb HTR-9 or anti-TNF-R2 mAb UTR-1). Inhibition of p38 significantly reduced PGE2 secretion in both RA- and OA-SFB (by about 95%). In the case of OA-SFB, the effects of both TNF-R1 and TNF-R2 were significantly inhibited by SB203580. Inhibition of ERK by U0126 or JNK by SP600125 did not significantly influence TNFα-induced PGE2 secretion (data not shown). In all cases, no significant differences were seen between RA and OA.
MMP-1 protein
Non-stimulated RA- and OA-SFB (n = 6 each) secreted large and comparable amounts of MMP-1 (table 1; fig 6A). Stimulation with TNFα significantly induced MMP-1 secretion (RA-SFB: 3.4-fold; OA-SFB: 4.4-fold; fig 6A), an effect exclusively mediated by TNF-R1. Inhibition of p38 significantly reduced TNFα-augmented MMP-1 secretion in OA-SFB (by about 35%), but not in RA-SFB (table 1; fig 6A). In general, inhibition of ERK by U0126 and JNK by SP600125 had no significant influence on TNFα-induced MMP-1 protein secretion, except for a significant reduction (by about 33%) of the TNFα-augmented MMP-1 protein secretion in RA-SFB by SP600125 (data not shown). In all cases, no significant differences were seen between RA and OA.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
MMP-1 and MMP-3 secretion in second passage SFB from patients with RA and OA after stimulation with TNFα, the agonistic anti-TNF-R1 mAb HTR-9, or the agonistic anti-TNF-R2 mAb UTR-1 (with or without inhibition of p38). In RA- and OA-SFB (n = 6 each), TNFα significantly induced the secretion of MMP-1 (A) and MMP-3 (B) compared with non-stimulated cells through TNF-R1. p38 inhibition reduced MMP-1 secretion solely in OA-SFB. Bars indicate means (SEM). *p<0.05 for the comparison of non-stimulated and stimulated cells; †p<0.05 for the comparison of cultures with or without SB203580; ‡p<0.05 for the comparison of RA- and OA-SFB.
MMP-3 protein
Stimulation with TNFα significantly induced MMP-3 secretion, an effect predominantly mediated by TNF-R1 (fig 6B). Inhibition of p38 (SB203580), ERK (U0126) or JNK (SP600125) did not significantly influence the TNFα-induced MMP-3 secretion (fig 6B; data not shown).
Correlations between the results and the clinical parameters of patients with RA and OA
Positive correlations were found between: (a) disease duration and MMP-1 protein levels in RA- and OA-SFB after stimulation with the agonistic anti-TNF-R1 mAb HTR-9 (r = 0.736; p = 0.006, n = 12); and (b) the number of positive ARA criteria and MMP-1 protein levels in RA-SFB after stimulation with TNFα (r = 0.926; p = 0.008, n = 6).
DISCUSSION
TNFα induces activation of the MAPK cascade in RA- and OA-SFB through TNF-R1
In agreement with previous data,13,31 phosphorylation of p38, ERK and JNK was observed in RA- and OA-SFB after TNFα stimulation. The phosphorylation of these signalling molecules was exclusively mediated through TNF-R1, underlining the central role of this receptor for MAPK signal transduction in SFB.
Notably, the ERK phosphorylation showed a delayed time course as compared with p38 and JNK activation. This observation indicates that the activation of individual MAPK signalling pathways after TNFα stimulation is regulated in a time-dependent manner, in analogy to recently reported data.13 Although the functional relevance of this time dependence is incompletely understood, the complete failure to influence pro-inflammatory/pro-destructive features of SFB by ERK inhibition may be caused by the fact that p38 and/or JNK activation precedes ERK activation.
TNFα reduces the proliferation of RA- and OA-SFB through TNF-R1
TNFα significantly downregulated the proliferation of RA- and OA-SFB, in sharp contrast to previous reports suggesting induction of fibroblast proliferation by TNFα.32,33 Possible explanations for these discrepancies include the differential usage of serum-free or serum-containing medium. On the other hand, the present results are in agreement with those obtained in normal lung fibroblasts, in which TNFα also reduced the proliferation rate.34 The suppression of proliferation was again exclusively mediated through TNF-R1, further emphasising the pivotal role of this TNF receptor.
Inhibition experiments showed that TNFα-mediated downregulation of proliferation is solely dependent on the p38 signal pathway. Inhibition of p38 had an effect upon TNFα/TNF-R1 stimulation, but did not significantly affect TNF-R2 stimulation. In addition, inhibition of ERK or JNK had no significant influence on the TNFα-induced downregulation of proliferation in RA- or OA-SFB. This shows that the p38 pathway has a central role for TNFα signal transduction through TNF-R1 and, at least for cell proliferation, has a differential importance for TNF-R1 and TNF-R2.
TNFα enhances the pro-inflammatory character of RA- and OA-SFB through TNF-R1
In good agreement with data on repeatedly-passaged cells,16,17 early-passage RA- and OA-SFB constitutively secreted IL6 and PGE2, without significant group differences. The secretion of these pro-inflammatory mediators was strongly upregulated by TNFα stimulation, an effect predominantly mediated by TNF-R1. Previous reports have shown a much weaker induction of IL6 protein by TNFα, possibly owing to the concurrent use of 10% FCS in cell culture (known to pre-stimulate cells).35 The present results, therefore, indicate that TNF-R1 plays a pivotal part also for the induction of pro-inflammatory mediators.
In contrast to previous reports in repeatedly passaged cells,17 however, there were no significant differences between RA- and OA-SFB for TNFα-induced IL6 secretion and TNF-R2-induced PGE2 secretion. Not only differences among patient cohorts but also different culture conditions (early-passage cells versus repeatedly passage cells), and possibly the use of different culture media (0.02% lactalbumin hydrolysate versus 5% FCS), have to be taken into consideration to explain these discrepancies.
Interestingly, in agreement with previous results,17 there was a moderate induction of PGE2 secretion after stimulation with the agonistic anti-TNF-R2 mAb. This indicates a possible role of TNF-R2 for the induction of pro-inflammatory mediators in RA- and OA-SFB, as also recently suggested in TNF-R2-overexpressing cell lines,36 which showed a higher secretion of pro-inflammatory IL6 after selective stimulation through TNF-R2.
In line with previously published data,21 there were quantitative differences between the effects of p38 inhibition on TNFα-induced IL6 secretion (about 50% reduction) and PGE2 secretion (90% reduction) in RA- and OA-SFB. A strong reduction of PGE2 secretion after p38 inhibition has also been reported in other cell types—for example, gastric cancer cells, human pulmonary artery smooth muscle cells, and chondrocytes,37–39 but not yet for SFB. Reduction of PGE2 release is attributed to lower COX-2 protein synthesis after p38 inhibition, and also to direct inhibition of the COX-2 activity by SB203580,40 a pyridinylimidazole compound with structural similarities to inhibitors of cyclo-oxygenase and lipoxygenases.
TNFα enhances the pro-destructive character of RA- and OA-SFB through TNF-R1
As previously reported for repeatedly passaged RA- and OA-SFB17 and skin fibroblasts,15 non-stimulated SFB secreted large amounts of MMP-1, without significant differences between RA and OA. Stimulation with TNFα clearly upregulated MMP-1 and MMP-3 secretion in both RA- and OA-SFB, an effect solely mediated by TNF-R1. These results further emphasise the importance of TNF-R1 for TNFα-induced regulation of pro-destructive molecules. In addition, significant positive correlations between MMP-1 protein levels after TNFα/TNF-R1 stimulation and the disease duration or number of positive ARA criteria suggest that the findings have clinical relevance.
Interestingly, TNFα/TNF-R1-induced MMP-1 protein secretion was significantly downregulated by p38 inhibition only in OA-SFB, but not in RA-SFB. Strikingly, p38 inhibition also failed to downregulate mRNA expression for MMP-1 in RA-SFB (data not shown). This suggests insensitivity of RA-SFB to p38 inhibition, possibly resulting in a partial resistance to the inhibition of pro-destructive functions.
The insensitivity of early-passage RA-SFB to p38 inhibition in the case of pro-destructive MMP-1 may be caused by several different underlying mechanisms. First, signalling pathways other than p38 (eg, NF-κB) may also have a central role in the TNFα-induced pro-destructive functions. Second, four different isoforms of p38 have been described (p38α, p38β, p38γ and p38δ), only two of which seem to be expressed in SFB (p38β and p38δ)41,42 and only two of which (p38α and p38β) are inhibited by SB203580.43–45 Therefore, a differential activation of the different p38 isoforms in RA-SFB might result in a partial unresponsiveness to p38 inhibition. Lastly, secondary mediators induced by TNFα—for example, IL6 or PGE2, may have a differential influence on the expression of MMP-1 and MMP-3 in RA- and OA-SFB.
Using either RA-SFB14 or gingival fibroblasts,46 other authors have described a significant reduction of TNFα-induced MMP-1 protein production by inhibition of p38. However, very high doses of the p38 inhibitors were required (RWJ 67657 or SB203580; 10 μmol/l in both cases), which bear the risk of non-specifically inhibiting kinases other than p38.45,47
In early-passage RA- and OA-SFB, TNFα-induced activation of MAPK cascades and pro-inflammatory/pro-destructive functions is predominantly mediated by TNF-R1 and, for proliferation and IL6/PGE2 secretion, exclusively regulated by p38. Strikingly, RA-SFB appear partially resistant to p38 inhibition of MMP-1 protein expression. Underlying structural or functional alterations of p38 and/or downstream components of the p38 pathway in RA-SFB may contribute to the pathogenesis or therapeutic sensitivity of RA, or both.
Acknowledgments
B Ukena, B Lanick and J Prechtel, Experimental Rheumatology Unit, Friedrich Schiller University, Jena, Germany, are gratefully acknowledged for technical assistance; Dr E Palombo-Kinne, for critical revision of the manuscript; Dr H Gellner, Helios Klinik, Blankenhain and Dr B Abendroth, Outpatient Practice for Rheumatic Diseases, Jena, Germany, for providing patient samples.
REFERENCES
Footnotes
-
Published Online First 11 January 2007
-
Elke Kunisch and Muktheshwar Gandesiri contributed equally to this work.
-
The study was supported by the German Federal Ministry of Education and Research (BMBF; grants FKZ 01ZZ9602, 01ZZ0105, and 010405 to RW Kinne, Interdisciplinary Centre for Clinical Research (IZKF) Jena, including a grant for junior researchers to E Kunisch; grants FKZ 0312704B and 0313652B to RW Kinne, Jena Centre for Bioinformatics; grant 01GS0413, NGFN-2 to RW Kinne), the German Research Foundation (DFG; grants KI 439/7-1 and KI 439/6-1 to RW Kinne), and a grant for the advancement of female scientists to E Kunisch (LUBOM Thuringia).
-
Competing interests: None.