Article Text

Abstract
Objectives To investigate efficacy and safety of the Janus kinase-1 inhibitor filgotinib in patients with active rheumatoid arthritis (RA) with limited or no prior methotrexate (MTX) exposure.
Methods This 52-week, phase 3, multicentre, double-blind clinical trial (NCT02886728) evaluated once-daily oral filgotinib in 1252 patients with RA randomised 2:1:1:2 to filgotinib 200 mg with MTX (FIL200 +MTX), filgotinib 100 mg with MTX (FIL100 +MTX), filgotinib 200 mg monotherapy (FIL200), or MTX. The primary endpoint was proportion achieving 20% improvement in American College of Rheumatology criteria (ACR20) at week 24.
Results The primary endpoint was achieved by 81% of patients receiving FIL200+ MTX versus 71% receiving MTX (p<0.001). A significantly greater proportion treated with FIL100+ MTX compared with MTX achieved an ACR20 response (80%, p=0.017) at week 24. Significant improvement in Health Assessment Questionnaire-Disability Index was seen at week 24; least-squares mean change from baseline was −1.0 and −0.94 with FIL200+MTX and FIL100+MTX, respectively, versus −0.81 with MTX (p<0.001, p=0.008, respectively). Significantly higher proportions receiving FIL200+MTX (54%) and FIL100+MTX (43%) achieved DAS28(CRP) <2.6 versus MTX (29%) (p<0.001 for both) at week 24. Hierarchical testing stopped for comparison of ACR20 for FIL200 monotherapy (78%) versus MTX (71%) at week 24 (p=0.058). Adverse event rates through week 52 were comparable between all treatments.
Conclusions FIL200+MTX and FIL100+MTX both significantly improved signs and symptoms and physical function in patients with active RA and limited or no prior MTX exposure; FIL200 monotherapy did not have a superior ACR20 response rate versus MTX. Filgotinib was well tolerated, with acceptable safety compared with MTX.
- arthritis
- rheumatoid
- therapeutics
- antirheumatic agents
Data availability statement
Anonymised individual patient data will be shared upon request for research purposes dependent upon the nature of the request, the merit of the proposed research, the availability of the data, and its intended use. The full data sharing policy for Gilead Sciences can be found at https://www.gilead.com/science-and-medicine/research/clinical-trials-transparency-and-data-sharing-policy.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Results from previous phase 3 studies and subsequent long-term follow-up of baricitinib, upadacitinib, and tofacitinib showed that treatment with small molecule Janus kinase (JAK) inhibitors with or without conventional synthetic disease-modifying antirheumatic drugs have a clinically meaningful benefit over methotrexate (MTX) monotherapy in reducing the signs and symptoms of rheumatoid arthritis, as well as radiographic progression, in MTX-naïve patients.
Safety signals such as increased risk of infections, venous thromboembolism, and major adverse cardiovascular event warrant careful evaluation of the risks and benefits of treatment, especially in the first-line treatment.
What does this study add?
This study is the first to evaluate filgotinib—a once-daily, oral, selective JAK-1 inhibitor—in patients with rheumatoid arthritis who had no or limited exposure to MTX.
Filgotinib, either alone or in combination with MTX, was found to have a clinically meaningful benefit over MTX monotherapy with an acceptable safety profile up to 52 weeks.
How might this impact on clinical practice or future developments?
Filgotinib in combination with MTX could be considered as a treatment option for patients with moderately or severely active rheumatoid arthritis who have limited or no previous exposure to MTX.
Introduction
Early control of rheumatoid arthritis (RA), including initiation of effective treatment as soon as possible, is critical to prevent disability, joint destruction, and decreased quality of life (QoL).1–4 For patients, the highest priority is returning to normality quickly.5 The European League Against Rheumatism (EULAR) and American College of Rheumatology (ACR) recommend initial therapy with methotrexate (MTX) alone or with glucocorticoids to induce remission or low disease activity by week 24, preferentially maintained over time.2 6 However, only approximately 30% of patients with RA achieve this objective with MTX monotherapy.7 8 Following treat to target approaches, including temporary glucocorticoids, this might increase to 60%–70%.9 Thus, alternative therapies providing rapid disease control are desirable. Such therapies will also benefit patients with seropositivity, elevated acute phase reactants, and early erosions, which, in a recently updated matrix, predict rapid radiographic progression.10
Filgotinib, an oral, selective Janus kinase (JAK)-1 inhibitor, was efficacious in phase 2 and phase 3 trials in patients with RA.11–14 In these trials, filgotinib 200 or 100 mg daily with conventional synthetic disease-modifying antirheumatic drugs (DMARDs) or as monotherapy demonstrated rapid, significant improvements in disease activity versus placebo.11–14 This phase 3 study (NCT02886728) evaluated the efficacy and safety of filgotinib with MTX or as monotherapy as compared with standard of care MTX monotherapy in patients with active RA with limited or no prior MTX exposure.
Methods
Study design and conduct
This 52-week, randomised, double-blind, active-controlled, phase 3 trial was conducted at 227 sites in 31 countries from 8 August 2016 to 5 October 2018. Patients were randomised 2:1:1:2 to receive filgotinib 200 mg with MTX (FIL200+MTX), filgotinib 100 with MTX (FIL100+MTX), filgotinib 200 mg (FIL200), or MTX. Patients provided written informed consent prior to screening.
Study participants
Eligible patients were ≥18 years with RA per 2010 ACR/EULAR criteria,15 limited (<3 doses ≤25 mg) or no prior MTX exposure, ≥6 swollen joint count of 66 joints (SJC66)), and ≥6 tender joints of 68 joints (TJC68)) at screening and day 1. At screening, eligible patients were required to have 1 of the following: seropositivity for rheumatoid factor and/or anticyclic citrullinated peptide or ≥1 radiographic erosion (centrally read); or serum C-reactive protein (CRP) ≥4 mg/L. Key exclusion criteria included previous use of JAK inhibitors or biological DMARDs. Prior or concomitant use of stably dosed hydroxychloroquine was allowed. Full inclusion/exclusion criteria are in online supplemental methods.
Supplemental material
Interventions
Filgotinib 100 or 200 mg were administered orally once daily. MTX was administered orally once weekly, starting with 10 mg/week and escalating to 15 mg at week 4 and 20 mg at week 8; in Japan, maximum 15 mg/week per local MTX prescribing practices. Local standard of care for folic acid supplementation was followed. After 4 weeks, a one-time 5 mg MTX dose reduction for intolerance was permitted if the dose remained ≥10 mg. Concomitant stable doses of non-steroidal anti-inflammatory drugs or glucocorticoids (≤10 mg/d prednisone/equivalent) were permitted.
Patients without 20% improvement from baseline in SJC66 and TJC68 at week 24, or at 2 consecutive visits from weeks 30 through 52, discontinued study drug and received standard of care, but continued study visits. Eligible patients completing the study could enter the long-term extension study (LTE) (NCT03025308); all others had a 4-week post-treatment visit.
Endpoints
The primary endpoint was proportion of patients receiving FIL200+MTX versus MTX achieving ACR2016 at week 24. Key secondary endpoints tested hierarchically at week 24 were ACR20 FIL100+MTX versus MTX, change from baseline in Health Assessment Questionnaire-Disability Index (HAQ-DI), proportion of patients achieving 28-joint Disease Activity Score (DAS28(CRP))<2.6, change from baseline in modified total Sharp/van der Heijde score (mTSS), Short Form 36–Physical Component Score (SF-36 PCS), and Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT-F; online supplemental figure S1).
Additional secondary and other endpoints evaluated over 52 weeks were the proportion of patients achieving ACR20/50/70, DAS28(CRP) <2.6, Simplified Disease Activity Index (SDAI)≤3.3, Clinical Disease Activity Index (CDAI) ≤2.8, and Boolean remission; and change from baseline in mTSS and its components, individual ACR components, DAS28(CRP), CDAI and SDAI. Radiographs were scored centrally as campaign A (radiographs taken at baseline and week 24) and campaign B (radiographs taken at baseline, week 24, and week 52 for patients who had images after week 24) by 2 blinded assessors and adjudicated by a third assessor, if necessary. Additional details are in online supplemental methods.
Safety was evaluated through adverse events (AEs) and laboratory assessments. All potential major adverse cardiovascular events (MACE) and thromboembolic events were adjudicated centrally by an independent committee. An independent data monitoring committee reviewed safety data periodically.
Statistical analysis
A sample size of 400 patients receiving FIL200+MTX and 400 receiving MTX monotherapy would provide 90% power at a two-sided α of 0.05 to test superiority of FIL200+MTX compared with MTX for change from baseline in mTSS at week 24, which is common in RA studies assessing structural damage.17–19 The total sample size was 1200 patients (400 patients for FIL200+MTX group, 200 patients for FIL100 +MTX group, 200 patients for FIL200 group, and 400 patients for MTX group). This sample size also provided >90% power to detect a 16% difference in ACR20 between patients receiving MTX (estimated response rate, 62%) versus any filgotinib treatment (78%) at a two-sided α of 0.05.
Type I error was controlled by hierarchical testing of primary and key secondary endpoints (online supplemental figure S1). The primary efficacy hypothesis was a superiority test (two-sided α=0.05) comparing ACR20 at week 24 for FIL200 +MTX versus MTX, using logistic regression analysis with treatment group and stratification factors, and non-responder imputation for missing data. Sequential testing for the primary and key secondary endpoints was implemented, and when a null hypothesis was not rejected, statistical significance was not considered for remaining hypotheses.
Efficacy and safety analyses were based on all randomised patients who received ≥1 dose of study drug. Efficacy analyses were based on on-treatment data. For binary endpoints, logistic regression was used with treatment and stratification factors (geographical region and seropositivity at screening) included in the model and non-responder imputation. Following treatment discontinuation, patient data were imputed as non-responses. Treatment effect on continuous endpoint change from baseline was evaluated with a mixed-effect model for repeated measures with treatment, visit, treatment by visit interaction, stratification factors, and baseline value as fixed effects and patient as a random effect. For structural endpoints, a linear mixed-effect model for change from baseline at week 24 (campaign A) and 52 (campaign A and B) and a generalised linear mixed-effect model for the proportion of patients with no radiographic progression (ie, Δ mTSS ≤0) at week 52 using campaign A and B data were employed.
Safety endpoints were analysed per treatment by number and percentage of patients with events for categorical values or summary statistics for continuous data. All statistical analyses were performed using SAS V.9.4 (SAS Institute).
Public involvement statement
The public was not involved in the design, conduct, or reporting of this research.
Results
Patients and disposition
Overall, 1252 patients were randomised, and 1249 received study treatment. In total, 90.5% patients completed study drug through week 24, and 82% through week 52 (figure 1). The largest proportion of patients enrolled from the USA (25.5%) and Eastern Europe (26.7%). Baseline demographic and disease characteristics were similar among treatment arms (table 1). At baseline, median disease duration was 0.4 years, and 77.2% of patients were DMARD naïve.

Study flow and patient disposition through 24 and 52 weeks. *Three patients were randomised (1 to FIL 200 mg+MTX and 2 to MTX monotherapy) but not dosed. FIL, filgotinib; MTX, methotrexate; QD, once daily; QW, once weekly.
Baseline demographics and clinical characteristics
Efficacy
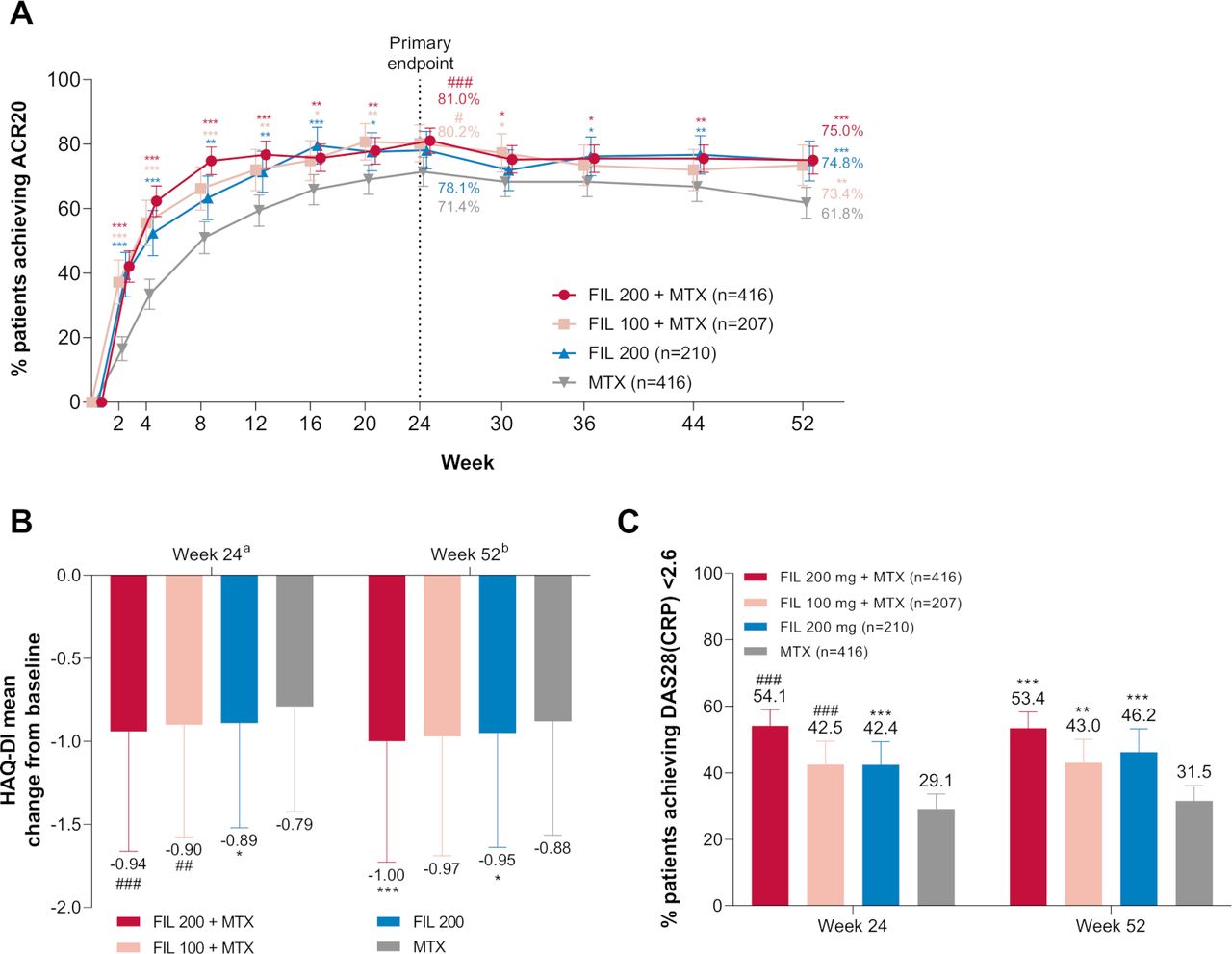
At week 24, the mean dose of MTX was 18 mg/week (online supplemental table S1). Significantly higher proportions of patients receiving FIL200+MTX (81%, p<0.001) and FIL100+MTX achieved ACR20 (80%, p=0.017) versus MTX (71%) at week 24 (figure 2A). Patients receiving either FIL200+MTX or FIL100+MTX had significant improvement in physical function HAQ-DI versus MTX at week 24; the least-squares mean (LSM) of the treatment difference in change in HAQ-DI from baseline versus MTX (95% CI) was −0.20 (−0.27,–0.12) (p<0.001) and −0.13 (−0.23,–0.03) (p=0.008) for FIL200+MTX and FIL100+MTX, respectively (figure 2B; table 2). Significantly higher proportions of patients receiving FIL200+MTX and FIL100+MTX achieved DAS28 (CRP) <2.6 versus MTX at week 24 (p<0.001 for all comparisons; figure 2C; table 2).

Primary, key secondary, and other secondary efficacy outcomes: (A) proportion of patients who achieved ACR20 over time; (B) change from baseline in HAQ-DI at week 24 and week 52; (C) proportion of patients achieving DAS28(CRP) <2.6 at week 24 and week 52. ###p<0.001; ##p<0.01; #p<0.05. The difference between filgotinib 200 mg and MTX for ACR20 at week 24 was not significant (p=0.058). ***Exploratory p<0.001; **exploratory p<0.01; *exploratory p<0.05, for supportive analysis without adjustment for multiplicity. an=372, 190, 185, and 370 for FIL200+MTX, FIL100+MTX, FIL200, and MTX, respectively. bn=332, 169, 171, and 307 for FIL200+MTX, FIL100+MTX, FIL200, and MTX, respectively. Error bars represent 95% CI for proportions of patients and SD for mean. For HAQ-DI, p values are based on least-squares mean difference versus MTX. Supporting values for (A) are shown in online supplemental table S3. ACR20, 20% improvement in American College of Rheumatology criteria; DAS28(CRP), 28-joint Disease Activity Score with C-reactive protein; FIL, filgotinib; HAQ-DI, Health Assessment Questionnaire-Disability Index; MTX, methotrexate.
Primary and key secondary efficacy outcomes at week 24
ACR20 for FIL200 (78%) at week 24 did not significantly differ from MTX (71%; p=0.058) (figure 2A). Therefore, exploratory p values without multiplicity adjustment are reported for all subsequent endpoints. At week 24, the LSM of the treatment difference in change in HAQ-DI from baseline for FIL200 versus MTX was −0.11 (figure 2B); 42% of patients receiving FIL200 achieved DAS28(CRP) <2.6 versus 29% with MTX.
At week 24, the LSM change from baseline in mTSS was 0.13 with FIL200+MTX, 0.13 with FIL100+MTX, and −0.13 with FIL200 versus 0.42 with MTX (figure 3A). Treatment with filgotinib or MTX improved health-related QoL, as measured with SF-36 PCS and FACIT-F. At week 24, change from baseline in SF-36 PCS was greater for FIL200+MTX and FIL100 +MTX versus MTX; and similar to MTX for FIL200, and improvements in FACIT-F scores were comparable across treatments (table 2).
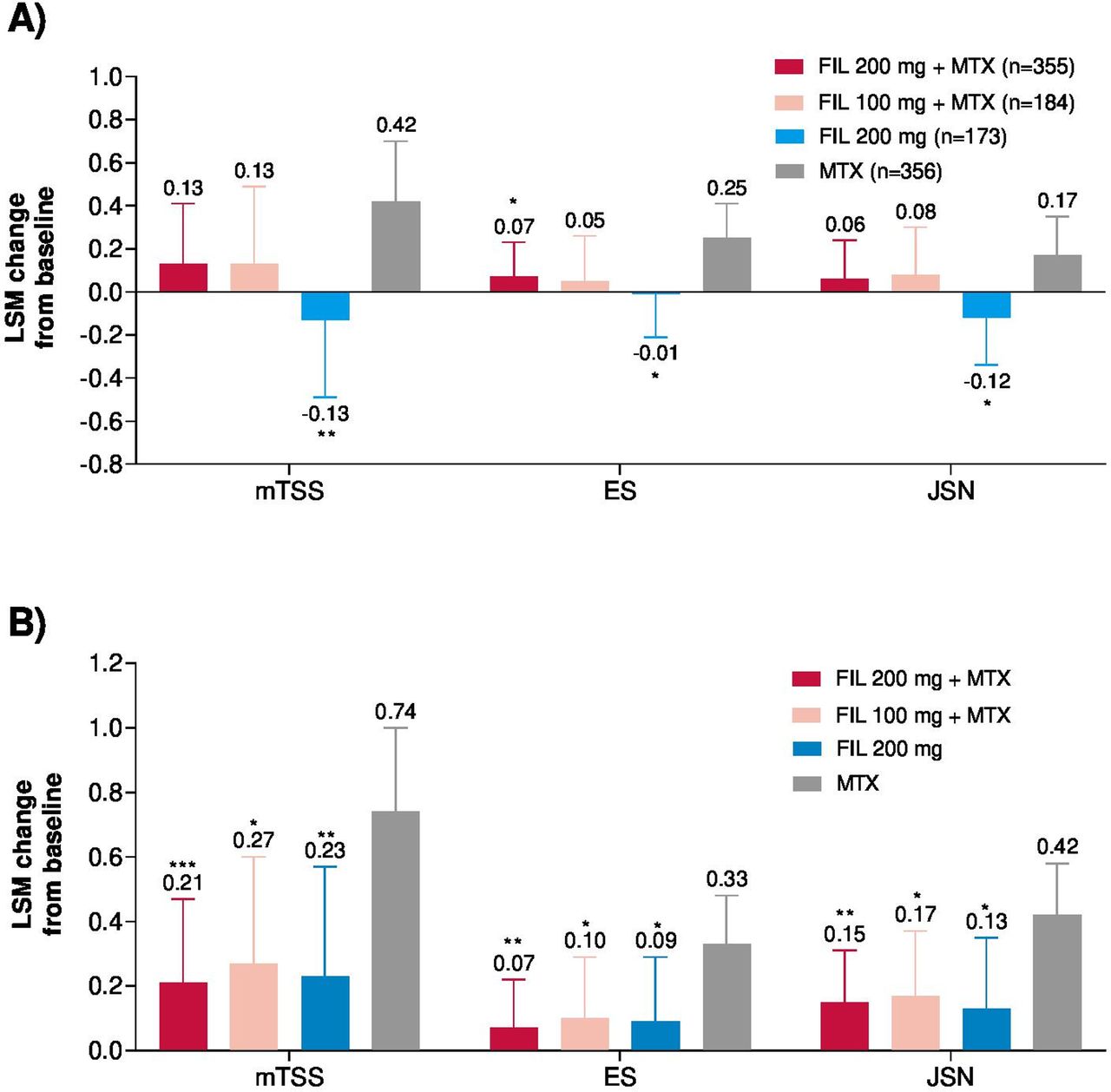
Change in modified total Sharp/van der Heijde score (mTSS) and components from baseline at (A) week 24 and (B) week 52. ***Exploratory p<0.001; **exploratory p<0.01; *exploratory p<0.05; for supportive analysis without adjustment for multiplicity. Error bars represent 95% CI. For mTSS, week 24 includes only data from campaign A and week 52 includes data from campaign A and B. Week 52 n values are not provided for mTSS change from baseline, as the analysis included both campaign A (through week 24) and campaign B (through week 52 including re-reading of baseline and week 24). ES, erosion score; FIL, filgotinib; JSN, joint space narrowing; LSM, least-squares mean; MTX, methotrexate.
At week 52, the LSM change from baseline in mTSS was 0.21 for FIL200+MTX, 0.27 for FIL100+MTX, and 0.23 for FIL200 versus 0.74 for MTX (figure 3B). More patients receiving any filgotinib treatment had no radiographic progression defined as Δ mTSS ≤0 at week 24 versus MTX (online supplemental table S2). The cumulative distribution plots of change from baseline in mTSS are shown in online supplemental figure S2.
At week 2, ACR20 response was achieved by 42.1%, 37.2%, and 39.5% of patients receiving FIL200+MTX, FIL100+MTX, and FIL200 versus 16.6% of patients receiving MTX (figure 2A). Proportions of patients who achieved ACR50/70 and DAS28 (CRP) <2.6 or remission at weeks 24 and 52 were higher with both doses of FIL+MTX or FIL200 mg versus MTX (figure 4). At week 24, all filgotinib groups resulted in greater improvement across components of ACR versus MTX (table 3).
Additional efficacy outcomes at week 24

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Proportion of patients achieving (A) ACR50 at weeks 24 and 52; (B) ACR70 at weeks 24 and 52; (C) remission at week 24; and (D) remission at week 52. ***Exploratory p<0.001; **exploratory p<0.01; *exploratory p<0.05; for supportive analysis without adjustment for multiplicity. ACR50/70, 50%/70% improvement in American College of Rheumatology criteria; CDAI, Clinical Disease Activity Index; FIL, filgotinib; MTX, methotrexate; SDAI, Simple Disease Activity Index.
Safety
The most common treatment-emergent AEs (TEAEs) occurring in >5% of patients were nausea, upper respiratory tract infection, nasopharyngitis, headache and hypertension. Serious TEAEs were reported for 6%, 6%, 8%, and 7% of patients receiving FIL200+MTX, FIL100+MTX, FIL200, and MTX, respectively.
Over 52 weeks, infection and serious infection rates were comparable for filgotinib versus MTX. Infections were reported for 36%, 37%, 36%, and 38% of patients receiving FIL200+MTX, FIL100+MTX, FIL200, and MTX, respectively; 1%, 1%, 2%, and 2% receiving FIL200+MTX, FIL100+MTX, FIL200, and MTX reported serious infections (table 4). The most common infections were upper respiratory tract infection, nasopharyngitis, urinary tract infection, and bronchitis. The most common serious infections were pneumonia, bronchitis, and sepsis. There were three opportunistic infections (FIL200+MTX, oesophageal candidiasis; MTX, Pneumocystis jirovecii pneumonia, pneumonia cryptococcal). There was one serious AE of non-disseminated herpes zoster in a Japanese patient in their 50s receiving FIL200 who required hospitalisation for intravenous antiviral therapy; other cases were uncomplicated.
Treatment-emergent adverse events and laboratory abnormalities up to week 52
Four deaths occurred: 3 in FIL200+MTX, 1 in FIL100+MTX. The causes of death included lupus myocarditis (possible overlapping systemic autoimmune disease), intracranial aneurysm, interstitial lung disease, and sudden cardiovascular death, which occurred 68 days after study drug discontinuation. A total of 9 MACEs were positively adjudicated during the study: 4 (1%) in FIL200+MTX, 1 (1%) in FIL100+MTX, 2 (1%) in FIL200, and 2 (1%) in MTX (table 4). All positively adjudicated MACE occurred in patients with cardiovascular risk factors. No venous thromboembolism (VTE) occurred with filgotinib; two positively adjudicated VTEs (pulmonary emboli) occurred with MTX treatment.
Malignancies, excluding non-melanoma skin cancer (NMSC) occurred in 4 (1%) patients receiving MTX (breast cancer, malignant melanoma, prostate cancer, and small cell lung cancer) and 1 (<1%) receiving FIL200+MTX (breast cancer). NMSC occurred in 2 (1%) patients receiving FIL200+MTX and 1 (<1%) receiving MTX. Diverticular gastrointestinal perforation occurred in 1 (<1%) patient receiving FIL200+MTX, non-steroidal anti-inflammatory drugs, and corticosteroids.
Laboratory abnormalities and TEAEs through 52 weeks are presented in table 4.
Mean haemoglobin increased slightly from baseline at week 52 with filgotinib (online supplemental figure S3). Mean decreases in platelets, neutrophils, and lymphocytes remained within normal range through week 52 for all treatments. There were no grade ≥3 increases in platelets. There was no temporal association between low lymphocytes and infection.
Grade 3 liver enzyme elevations were more frequent versus MTX with FIL200+MTX and FIL100+MTX, but comparable for FIL200 and MTX (table 4). However, no case satisfied Hy’s law to suggest drug-induced hepatocellular injury. Grade 3 creatinine increase was reported with MTX; none occurred with filgotinib. Grade 3 creatine kinase (CK) elevation incidence was similar with filgotinib versus MTX; grade 4 elevation occurred only with filgotinib. These events were transient, without symptoms of muscle toxicity or rhabdomyolysis. One patient receiving FIL200 discontinued due to asymptomatic grade 3 CK elevation. Cholesterols increased by a mean (SD) of 17 (28), 12 (28), and 14 (29) mg/dL for low-density lipoprotein (LDL) cholesterol and 11 (15), 6 (14), and 12 (15) mg/dL for high-density lipoprotein (HDL) cholesterol with FIL200+MTX, FIL100+MTX, and FIL200, respectively; however, LDL/HDL ratios remained stable (table 4).
Discussion
Early diagnosis and treatment are desirable to achieve RA treatment goals: induction of remission, maintenance of physical function, and maximisation of patient QoL.2 6 Although treatment with all filgotinib regimens and MTX in the current study were effective in improving disease activity, physical function, and patient reported outcomes, patients who received FIL200+MTX or FIL100+MTX achieved a superior ACR20 response and improvements in HAQ-DI and DAS28(CRP) <2.6 at week 24 compared with MTX. However, the proportion of patients achieving ACR20 at week 24 treated with FIL200 monotherapy did not attain statistical significance versus MTX in the hierarchical testing scheme.
The clinical benefits achieved in the study were considered alongside the potential risks of all treatment regimens. Over 52 weeks, rates of serious AEs, infections, serious infections, opportunistic infections, herpes zoster, MACE and malignancies were similar between filgotinib and MTX arms. No VTEs were observed in patients receiving filgotinib. Three deaths occurred in patients receiving filgotinib and an additional death occurred more than 2 months after cessation of filgotinib. There was no pattern in specific cause of death, and events were consistent with common causes of mortality in RA apart from the event of lupus myocardiopathy.20
Liver enzyme elevations were more commonly observed with filgotinib in combination with MTX than with either filgotinib monotherapy or MTX. However, no reported case satisfied Hy’s law to suggest drug-induced liver injury. Filgotinib, either as a monotherapy or with MTX, was associated with decreases in neutrophil, lymphocyte, and platelet counts, and increases in lipids and CK. These results were similar to prior filgotinib trials.11–13 MTX monotherapy was also associated with decreases in neutrophil, lymphocyte, and platelet counts; such decreases are an expected feature of successful reduction in RA disease activity. The longer term safety of filgotinib is being evaluated in two ongoing LTE trials (NCT02065700; NCT03025308).
Limitations of this study include that the design precluded adjusting the MTX dose following the initial 8-week period, increasing the dose >20 mg/week, or switching to an injectable formulation. Additionally, patients receiving concomitant glucocorticoids were required to receive a stable dose, precluding use as short-term bridging therapy. The lack of a placebo group might have increased the response in all treatment groups due to expectation bias. Nevertheless, the responses observed with filgotinib are consistent with other studies of filgotinib.11–13 The inability to demonstrate superiority of FIL200 monotherapy versus MTX for ACR20 at week 24 precluded further claims of statistical significance for other clinically important endpoints, such as physical function and remission, for which FIL200 appeared to have benefit over MTX. Finally, the low progression rate of structural damage compromised the ability to demonstrate a benefit between filgotinib arms versus MTX.
In early RA, separation between two active medications can be observed based on the proportion of patients achieving the clinically meaningful threshold such as the ACR20 but also by the depth of response seen by those on treatment. Thus, two medications can show similar proportions of ACR20 responses, and their true difference in efficacy is only revealed when assessing ACR50 and ACR70 responses.21 Although ACR20 was chosen as the primary endpoint, other endpoints of clinical interest were also evaluated. All filgotinib treatments achieved numerically better disease control versus MTX alone for higher hurdle endpoints such as ACR50/70 and remission.
Early intensive treatment aims to limit both disease activity—ideally inducing disease remission—and progression of structural damage that could lead to loss of function and disability in the long term. Despite aiming to enrich the study with patients at risk for structural progression, the proportion of patients in whom structural damage progressed was low. While longer duration of follow-up may be necessary to demonstrate the impact of filgotinib on irreversible joint damage, overall, there was less progression of structural damage with all filgotinib treatments versus MTX alone at week 52 and with FIL 200 mg at week 24. These comparisons were not adjusted for multiplicity, and thus no final conclusion can be drawn.
As current guidelines recommend early initiation of DMARDs after RA diagnosis, it is important to note that 34% of the patients enrolled in the study had disease duration >1 year with no DMARD therapy prior to study enrolment, which may limit the generalisability of our findings. However, in sensitivity analyses in which patients were stratified by disease duration of <1 or>1 year (data not shown), results were similar regardless of disease duration for all endpoints evaluated.
In patients with active RA who had limited or no prior MTX exposure, filgotinib with MTX reduced signs and symptoms of RA and improved physical function, and these effects were sustained through 52 weeks. Both filgotinib doses were well tolerated with an acceptable safety profile. Given the high-hurdle outcomes at week 52, an initial dose of filgotinib 200 mg eventually tapering to 100 mg in patients with good disease control at 1 year may be considered in clinical practice. An additional study evaluating early, temporary use of JAK-inhibitors or bDMARDs in patients with a suboptimal early response to initial MTX and tapering glucocorticoids is needed to assess potential benefits of this strategy.
Data availability statement
Anonymised individual patient data will be shared upon request for research purposes dependent upon the nature of the request, the merit of the proposed research, the availability of the data, and its intended use. The full data sharing policy for Gilead Sciences can be found at https://www.gilead.com/science-and-medicine/research/clinical-trials-transparency-and-data-sharing-policy.
Ethics statements
Ethics approval
Institutional review boards or ethics committees approved the protocol; the study was conducted in accordance with the Declaration of Helsinki and International Council for Harmonisation Good Clinical Practice guidelines.
Acknowledgments
Medical writing support was provided by Kathleen Pieper, PhD, of AlphaScientia, LLC, and funded by Gilead Sciences.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Handling editor Josef S Smolen
Presented at This work was presented at the 2019 European League Against Rheumatism annual meeting in Madrid, Spain (Westhovens, et al. Ann Rheum Dis. 2019;78[Suppl2]:259–60) and the 2020 virtual European League Against Rheumatism annual meeting (Westhovens, et al. Ann Rheum Dis. 2020;79[Suppl1]:1019–20).
Contributors FM, NM, JSS, and CT contributed to the study design. BB, NM, and JSS oversaw the conduct of the study. RW, WFCR, DWTC, WS, JK, AC, ODM, TA, and GRB participated in the conduct of the study. ZY and YG performed the statistical analysis. All authors contributed to the interpretation of the results, critically reviewed the manuscript during writing, and approved of the final draft for submission.
Funding Funding for the study was provided by Gilead Sciences, Inc.
Competing interests RW reports grant/research support from and serving as a consultant for Celltrion, Galapagos, and Gilead Sciences. WFCR reports serving as a consultant for Gilead Sciences. DvdH reports serving as a consultant for AbbVie, Amgen, Astellas, AstraZeneca, Bristol-Myers Squibb, Boehringer Ingelheim, Celgene, Cyxone, Daiichi Sankyo, Eisai, Eli Lilly and Co., Galapagos, Gilead Sciences, GlaxoSmithKline, Janssen, Merck, Novartis, Pfizer, Regeneron, Roche, Sanofi, Takeda, and UCB Pharma; and is director of Imaging Rheumatology BV. DWTC reports receiving grants from AbbVie, Gilead Sciences, Pfizer, and Sanofi; and serving as a consultant for AbbVie and Pfizer. WS reports receiving consulting fees from Janssen Research and Development, and research support from GlaxoSmithKline. JK reports research grants paid to his institution from AbbVie, Genentech, Gilead Sciences, Pfizer, and UCB; and has received personal fees from AbbVie, Alvotech,Suisse AG, Amgen, Boehringer Ingelheim GmbH, Celltrion Healthcare Co., Janssen Biotech, Merck Sharp & Dohme Corp., Mylan, Novartis AG, Pfizer, Samsung Bioepis, Sandoz, and UCB. AC reports no conflicts of interest. BB, FM, ZY, and YG are shareholders and employees of Gilead Sciences. CT is a shareholder and employee of Galapagos NV. JSS is a shareholder and former employee of Gilead Sciences; and current employee and shareholder of Pandion Therapeutics. NM is a shareholder and former employee of Gilead Sciences, and current employee of Ichnos Sciences. AJ is a shareholder and former employee of Gilead Sciences. ODM reports serving on a speaker’s bureau for Americas Health Foundation, Amgen, and Pfizer. RBML reports serving as a consultant for AbbVie, AstraZeneca, Bristol-Myers Squibb, Eli Lilly and Co., Galapagos NV, Novartis, Pfizer and UCB. TA reports grant/research support from AbbVie, Alexion, Astellas, Bristol-Myers Squibb, Chugai Pharmaceutical Co., Daiichi Sankyo Co., Eli Lilly Japan K.K., Mitsubishi Tanabe Pharma Co., and Pfizer; and has served as a consultant for AbbVie, Chugai, Daiichi Sankyo Co., Eli Lilly Japan K.K., Gilead Sciences, Pfizer, and UCB Japan Co.; and has served on a speakers bureau for AbbVie, Astellas, Bristol-Myers Squibb Co., Chugai Pharmaceutical Co., Daiichi Sankyo Co., Eisai Co., Eli Lilly Japan K.K., Mitsubishi Tanabe Pharma Co., Otsuka Pharmaceutical Co., Pfizer, Takeda Pharmaceutical Co., and UCB Japan Co. GRB reports serving as a consultant and on a speaker’s bureau for AbbVie, Eli Lilly and Co., Pfizer, and Gilead Sciences.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.