Article Text

Abstract
Objective To evaluate efficacy and safety of ustekinumab in patients with ankylosing spondylitis (AS).
Methods In this prospective, open-label, single-arm, proof-of-concept clinical trial (ClinicalTrials.gov identifier NCT01330901), ustekinumab in a dose of 90 mg was administered subcutaneously at baseline, week 4 and week 16 in 20 patients with active AS. Eligible patients were required to have a diagnosis of AS according to the modified New York criteria and an active disease defined as a Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) score of ≥4 despite previous non-steroidal anti-inflammatory drug (NSAID) treatment. The primary study endpoint was the proportion of patients reached the Assessment of SpondyloArthritis International Society 40 (ASAS40) response at week 24.
Results At week 24, ASAS40 response was reached by 65% of the patients. ASAS20, ASAS5/6 and ASAS partial remission were observed in 75%, 50% and 30% of the patients, respectively. A ≥50% improvement of the BASDAI (BASDAI50) occurred in 55% of the patients. A total of 50% and 20% of the patients achieved the AS Disease Activity Score (ASDAS) clinically important improvement and major improvement, respectively. At week 24, 35% of the patients had an ASDAS inactive disease (ASDAS <1.3). Significant improvement of other patient-reported outcome parameters and active inflammation as detected by MRI as well as significant reduction of NSAIDs intake occurred during the treatment. Clinical response correlated with reduction of active inflammation on MRI and of serum C reactive protein level. Overall, ustekinumab was well tolerated.
Conclusions In this prospective, open-label, proof-of-concept clinical trial, ustekinumab treatment was associated with a reduction of signs and symptoms in active AS and was well tolerated.
- Ankylosing Spondylitis
- Treatment
- Spondyloarthritis
Statistics from Altmetric.com
The therapeutic options in ankylosing spondylitis (AS) with predominant spinal manifestations are limited and confined to non-steroidal anti-inflammatory drugs (NSAIDs) and, if this treatment fails, to tumour necrosis factor (TNF) α blockers.1 ,2 This is in contrast to other inflammatory rheumatic diseases, such as rheumatoid arthritis, where a variety of treatment options are available. Indeed, several conventional disease-modifying antirheumatic drugs (DMARDs) have been investigated in AS,3–5 but have failed so far. Similarly, the biologics anakinra,6 abatacept7 and monoclonal antibodies directed against the interleukin (IL)-6 receptors8 ,9 did not show any efficacy in patients with active AS.
Regarding TNFα blocking therapy, up to 40% of the patients treated in the pivotal studies were non-responders, as defined by the rate of Assessment of SpondyloArthritis International Society 20 (ASAS20) response.10–13 This failure rate is slightly reduced if switching to a second TNFα blocker is also taken into account in case of failure.14 Thus, new therapeutic options are urgently needed for patients with active AS.
Recent findings suggest that IL-23 might have a role in the pathogenesis of AS. A polymorphism within the IL-23 receptor was found to affect susceptibility to AS.15 ,16 Interestingly, similar associations were reported for psoriasis17 and inflammatory bowel disease.18 Furthermore, we just reported that IL-23 is expressed in the subchondral bone marrow and fibrous tissue replacing bone marrow in facet joints of patients with AS,19 and elevated IL-23 concentrations were found in peripheral blood and synovial fluid from AS patients.20 ,21 IL-23 plays a key role in driving CD4 memory T cells to produce pro-inflammatory cytokines such as IL-1722 ,23 and a recent small controlled study in AS found that a monoclonal antibody directed against the IL-17A cytokine, secukinumab, showed some efficacy in comparison with a control group, with an ASAS40 response rate of 30% at week 6 in the secukinumab treated group.24 Finally, IL-23 had a crucial role both in inflammation and in new bone formation in an animal model with some features resembling those of spondyloarthritis.25
Ustekinumab is a fully human IgG1κ monoclonal antibody that binds with high affinity and specificity to the p40 protein subunit of the human cytokines IL-12 and IL-23. Ustekinumab inhibits the activity of human IL-12 and IL-23 by preventing these cytokines from binding to their IL-12Rβ1 receptor protein expressed on the surface of immune cells. Previously, ustekinumab has been shown to be effective in the treatment of psoriasis26–28 and psoriatic arthritis29 ,30 and is currently under investigation in a phase III trial in Crohn's disease31—conditions sharing common pathogenetic mechanism with AS.
The aim of this proof-of-concept trial (UsTekinumab for the treatment Of Patients with active Ankylosing Spondylitis - TOPAS) was to investigate efficacy and safety of ustekinumab in patients with active AS.
Methods
Study design
In this prospective, open-label, proof-of-concept clinical trial (ClinicalTrials.gov identifier NCT01330901), ustekinumab was administered subcutaneously in a dose of 90 mg at baseline, week 4 and week 16 in 20 eligible patients with active AS. After week 16, patients continued in a 12-week follow-up period till week 28.
Inclusion and exclusion criteria
Eligible patients were required to have a diagnosis of AS according to the modified New York criteria32 and an active disease defined as a Bath Ankylosing Spondylitis Disease Activity Index (BASDAI)33 score of ≥4 despite concomitant treatment with an NSAID (or without NSAIDs in case of intolerance/contraindication) at screening and history of an inadequate response to ≥2 NSAIDs or NSAID intolerance/contraindication. Current NSAID therapy had to be stable for at least 2 weeks prior to baseline. Concomitant treatment with glucocorticoids (≤10 mg prednisone equivalent per day) and methotrexate (but not with other DMARDs) was permitted, but had to be stable 4 weeks prior to baseline. The main exclusion criteria were history of non-response to previous TNFα blocking therapy (patients who discontinued TNFα blockers for reasons other than lack of efficacy were allowed to participate), uncontrolled concomitant diseases, pregnancy, and clinical and laboratory abnormalities.
Outcome assessments
The primary end point was a 40% improvement in disease activity at week 24 according to the ASAS criteria (ASAS40).34
Secondary outcome parameters were ASAS20 response,35 ASAS5/6 response,34 ASAS partial remission,35 50% improvement of the BASDAI (BASDAI50), clinically important improvement (change score ≥1.1) and major improvement (change score ≥2.0) of the C reactive protein (CRP) based ASAS endorsed AS Disease Activity Score (ASDAS),36 ASDAS inactive disease state (status score <1.3),37 the mean improvement in ASDAS, BASDAI, the Bath Ankylosing Spondylitis Functional Index (BASFI),38 the Bath Ankylosing Spondylitis Metrology Index (BASMI),39 patient global, physician global, general and nocturnal pain on the numeric rating scale, CRP level, erythrocyte sedimentation rate (ESR), improvement of quality of life and disability measurements (EQ-5D40 and AS quality of life (ASQoL)41 scales), achievement of the patient acceptable symptom state42 and of the physician acceptable symptom state at week 24. In patients with peripheral manifestations, the swollen joint count and improvement of enthesitis, as assessed by the Maastricht Ankylosing Spondylitis Enthesitis Score (MASES),43 and the Spondyloarthritis Research Consortium of Canada (SPARCC) enthesitis index,44 were analysed as additional secondary outcome parameters. NSAID intake was flexible after baseline and counted at every study visit starting from baseline through week 24 as recommended by ASAS.45 Based on the collected data related to the NSAID dose and intake frequency, the ASAS NSAIDs intake score was calculated.45
MRI of the sacroiliac joints and of the spine was performed by the short τ inversion recovery sequence at baseline and at week 24 (or earlier in case of study discontinuation prior to week 24). MRI images were scored independently by two trained readers (DP and KGH) in a concealed and randomly selected order, blinded for all clinical data. Active inflammation (osteitis/bone marrow oedema) was scored according to the Berlin scoring system.46
Safety outcome parameters include the percentage of patients experiencing adverse events (AEs) and number of AEs/serious AEs from baseline through week 28.
Statistics
For the sample size calculation, an ASAS40 response of 40% in the ustekinumab group was anticipated. Based on the results of the placebo-controlled trials,10–13 an ASAS40 placebo response of 14% was assumed. Based on these assumptions, a sample size of 20 patients in the treatment group was chosen in order to achieve a 70% power of an exact one sample binomial test comparing the response in the treatment group with the anticipated ASAS40 placebo response.
Efficacy analysis was performed using the intention-to-treat (ITT) population, which include all patients who received at least one dose of study medication. In the safety analysis, all patients receiving at least one dose of the study drug were included. The primary and secondary endpoints at week 24 were investigated by means of descriptive statistics in the whole sample. Response rates are given as numbers of responders, percentages and the 95% CI of the percentages. The outcome of metrical scales, such as BASDAI and BASFI, are given as means and SDs. Osteitis MRI scores were calculated separately for the sacroiliac joints (range 0–24) and for the spine (range 0–69). Mean score values of two readers are reported. Agreement between readers was estimated by the intraclass correlation coefficient (ICC). The paired sample t test was used to compare changes between baseline values and values after treatment. The Mann–Whitney U test was used to compare parameter values between responders and non-responders. In the ITT analysis, the last observation carried forward method was applied to calculate the outcome parameters in all patients who received at least one dose of ustekinumab.
Results
Efficacy assessment
In total, 22 patients with active AS were screened for this study; 20 of them were considered to be eligible and were treated with ustekinumab. A total of three patients discontinued the study prematurely (two patients after week 8 and one patient after week 4) due to lack of efficacy (all were counted as non-responders in the analysis population), while 17 patients finished the study according to the protocol. The baseline characteristics of the study population are presented in table 1.
Main baseline demographic and clinical characteristics of patients with active AS (n=20) treated with ustekinumab in the TOPAS trial
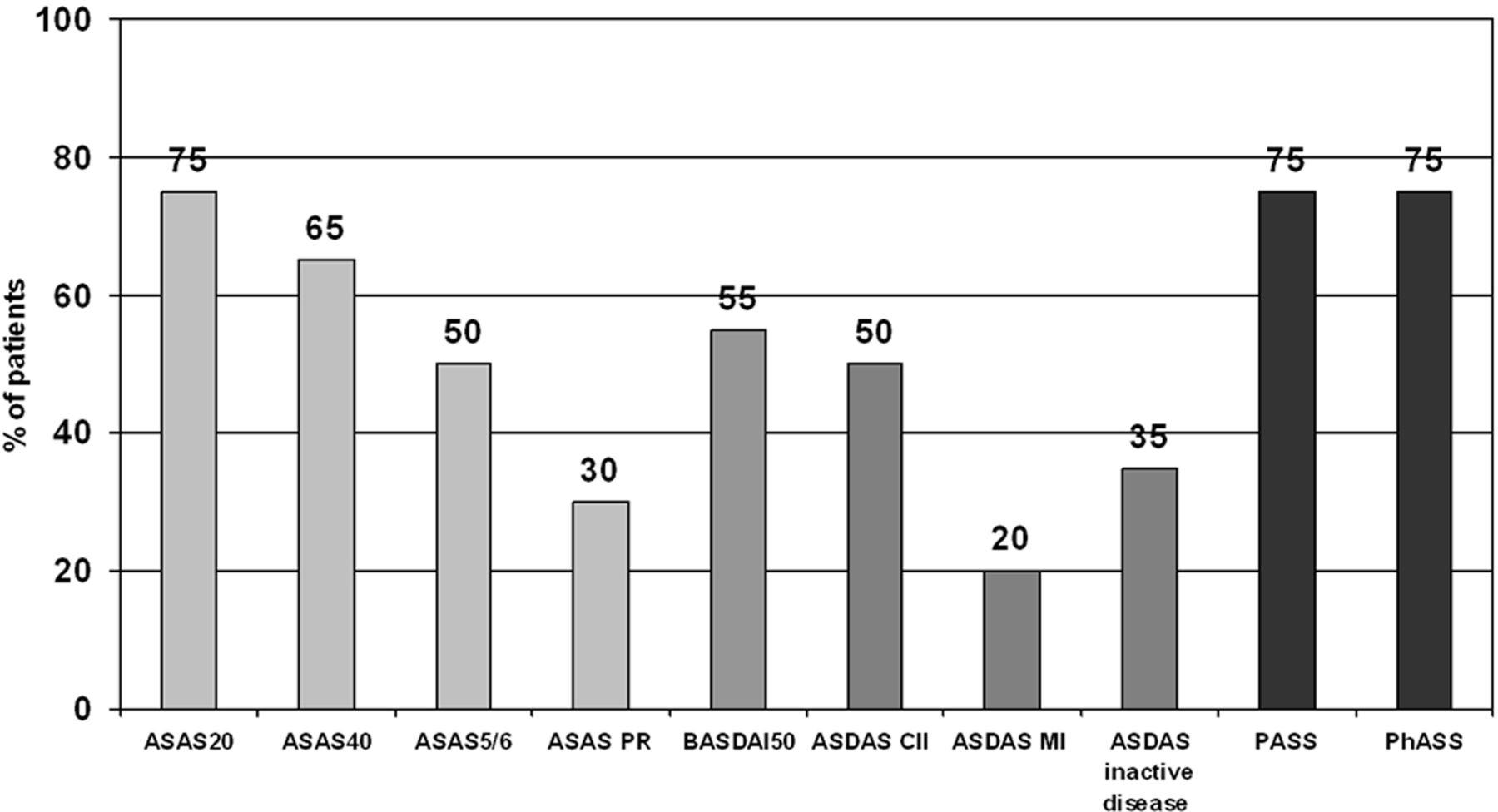
In total, 13 out of 20 patients (65%, 95% CI 41% to 85%) achieved the primary endpoint (the ASAS40 response—at week 24) (figure 1). Regarding secondary outcome parameters, 15 patients (75%, 95% CI 53% to 90%) achieved the ASAS20 response, 10 patients (50%, 95% CI 29% to 71%) ASAS5/6 response and six patients (30%, 95% CI 14% to 53%) were even in ASAS partial remission at week 24. At least 50% improvement of the BASDAI was achieved by 11 patients (55%, 95% CI 32% to 77%). Regarding ASDAS responses at week 24, 50% (95% CI 29% to 71%) of the treated patients achieved a clinically important improvement (ASDAS absolute improvements by ≥1.1 as compared with baseline), 20% (95% CI 7% to 41%) achieved a major improvement (ASDAS improvement ≥2.0) and 35% (95% CI 15% to 59%) of the patients reached inactive disease (ASDAS <1.3) at week 24. Interestingly, identical high rates of patient and physician acceptable symptom state at week 24—75% (95% CI 53% to 90%)—were similar to the magnitude of the ASAS20 response.

Response rates to ustekinumab at week 24 in the TOPAS trial (n=20). ASAS, Assessment of SpondyloArthritis International Society; ASAS PR, ASAS partial remission; ASDAS, AS Disease Activity Score; ASDAS CII, ASDAS clinically important improvement (change score ≥1.1 from baseline); ASDAS MI, ASDAS major improvement (change score ≥2.0 from baseline); BASDAI, Bath Ankylosing Spondylitis Disease Activity Index; PASS, patient acceptable symptom state; PhASS, physician acceptable symptom state.
All continuous parameters related to disease activity (ASDAS, BASDAI, patient and physician global, general and nocturnal pain), axial mobility (BASMI, chest expansion), function, disability and quality of life (BASFI, EQ-5D, ASQoL) improved substantially and significantly at week 24 compared with baseline (table 2). Clinical improvement occurred after the first ustekinumab injection and reached a statistical significance: for instance, at week 2 for BASFI, and week 8 for BASDAI and ASDAS (figure 2). However, there was a further continuous improvement until the end of the study.
Changes in the clinical, laboratory and imaging parameters over 24 weeks in patients with active AS (n=20) treated with ustekinumab in the TOPAS trial

Mean values of the BASDAI, BASFI and ASDAS scores over time in patients with active AS (n=20) treated with ustekinumab. AS, ankylosing spondylitis; ASDAS, Ankylosing Spondylitis Disease Activity Score; BASDAI, Bath Ankylosing Spondylitis Disease Activity Index; BASFI, Bath Ankylosing Spondylitis Functional Index.
Enthesitis, measured by the MASES and SPARCC enthesitis indices at week 24, was not significantly different from baseline (table 2). There was no effect on the number of swollen joints (table 2), which can possibly be explained by an insufficient number of patients with peripheral arthritis (n=1).
Among the entire group (n=20), there was no improvement in the level of CRP (and to a lesser extent, ESR) at week 24 (table 2). However, there was a clear improvement of CRP at week 24 in patients who demonstrated a clinical response (ie, ASAS40, n=13) at this time point, while a worsening of CRP level was observed in those without ASAS40 response (n=7): −1.1±7.5 mg/L versus +3.3±3.5 mg/L, respectively; p=0.008. This was even clearer if a 50% BASDAI improvement was used as a response criterion: CRP change −3.3±2.6 mg/L in responders (n=11, figure 3A) versus +5.0±7.3 mg/L in non-responders (n=9, figure 3B), respectively; p=0.001.

Changes in C reactive protein levels in AS patients with (A) and without (B) clinical response (BASDAI50) to ustekinumab over 24 weeks of treatment. AS, ankylosing spondylitis; BASDAI50, a 50% improvement of the Bath Ankylosing Spondylitis Disease Activity Index.
The full sets of MRIs (baseline and follow-up—all but one performed at week 24, in one case MRI was performed after early study discontinuation at week 8) were available for 17 patients (13 ASAS40 responders and four non-responders). The ICC for osteitis scores for the sacroiliac joints and for the spine were 0.94 (95% CI 0.86 to 0.98) and 0.64 (95% CI 0.30 to 0.84), respectively, at baseline, and 0.84 (95% CI 0.58 to 0.94) and 0.67 (95% CI 0.30 to 0.87), respectively, at follow-up. There was a significant reduction of active inflammation (osteitis) on MRI at week 24 as compared with baseline in both sacroiliac joints (osteitis change score −2.2±3.8 corresponding to 41% reduction) and spine (osteitis change score –1.2±2.3 corresponding to 31% reduction; table 2). Again, reduction of active inflammation after 24 weeks was more prominent and statistically significant in patients with clinical response (ie, ASAS40): osteitis change score in the sacroiliac joints was −3.1±3.8 in responders as compared with +0.6±1.3 in non-responders, p=0.015; similarly, osteitis change score in the spine was −1.9±1.9 in responders as compared with +1.0±2.4 in non-responders, p=0.023. Figure 4 represents examples of reduction of active inflammation in the sacroiliac joints and spine under ustekinumab treatment.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Reduction of active inflammation in the sacroiliac joints and spine in patients with ankylosing spondylitis treated with ustekinumab as detected by MRI. (A and B) MRI of the sacroiliac joints (STIR) at baseline and week 24 of a 23-year-old patient who was an ASAS40 responder at week 24. Arrows indicate active inflammation (osteitis) at baseline and nearly complete resolution of inflammation at week 24. (B and C) MRI of the lumbar spine (STIR) at baseline and week 24 of a 40-year-old patient who was an ASAS40 responder at week 24. Arrows indicate active inflammation (osteitis) at baseline and nearly complete resolution of inflammation at week 24. ASAS40, Assessment of SpondyloArthritis International Society 40; STIR, short τ inversion recovery.
Patients who achieved the primary outcome (ASAS40 response, n=13) had higher levels of inflammation as detected by MRI in the sacroiliac joints at baseline (6.7±4.9 in responders vs 2.0±1.7 in non-responders; p=0.030) and showed trends towards younger age (36.7±10.5 years in responders vs 39.0±12.1 years in non-responders; p=0.643), shorter symptom duration (12.5±10.6 years in responders vs 15.0±10.8 years in non-responders; p=0.536), lower level of functional limitations at baseline as measured by the BASFI (5.0±1.9 in responders vs 5.9±2.0 in non-responders; p=0.485), higher level of CRP (7.4±6.8 mg/L in responders vs 6.6±6.3 mg/L in non-responders; p=0.241) and higher level of spinal inflammation on MRI (4.9±3.6 in responders vs 3.6±4.1 in non-responders; p=0.241).
NSAID intake was reduced substantially over 24 weeks of treatment with ustekinumab. At baseline, 17 patients (85%) took NSAIDs, and after 24 weeks 13 patients (65%) were on NSAID treatment. The majority of patients who remained on NSAIDs at week 24 were able to reduce the dose and/or frequency of NSAID intake as reflected by reduction of the ASAS NSAID intake score from 68.7±37.9 at baseline to 33.3±33.6 at week 24; p=0.008.
Safety assessment
In general, ustekinumab was well tolerated. A total of 92 AEs were observed in the study (all of mild or moderate severity); at least one AE was reported in 19 out of 20 patients, but no drop-outs occurred because of AEs. Only one serious AE occurred: worsening of AS-related back pain, which resulted in hospitalisation (but not to a study discontinuation). The most common reported AEs were upper respiratory tract infections in 14 cases (15%), followed by rhinitis in seven cases (8%), abdominal pain/discomfort in seven cases (8%), headache in seven cases (8%) and diarrhoea in four cases (4%). There were no injection site reactions. No serious infections, opportunistic infections, cases of tuberculosis, cases of malignancies or deaths were observed.
Discussion
In this prospective, open-label, single-arm, proof-of-concept trial, we showed that patients with active AS treated with ustekinumab achieved high response rates after 6 months, with 65% of the patients reaching the primary outcome parameter of ASAS40 and 55% reaching BASDAI50 response. For comparison, in other open-label trials with a similar study design, the ASAS 40 response rate was reached in only 20%, 13% or 10% of patients treated with the IL-1 receptor antagonist anakinra,6 the T cell response modulator abatacept7 or with subcutaneous methotrexate in weekly doses up to 20 mg,4 respectively. In a recent placebo-controlled trial, the ASAS40 response was only 11.8% in AS patients treated with the IL-6 receptor antagonist tocilizumab.9 For further comparison, the ASAS40 response was reached by about 40% of patients treated with TNFα blockers in the pivotal trials.10–13 Thus, with the limitation of any treatment trial which is not blinded and which does not include a placebo group, our data suggest that ustekinumab is a promising drug for the treatment of active AS and justifies the conduct of a larger placebo-controlled trial.
Interestingly, in about a third of non-responders, defined by ASAS40 response, a worsening of the continuous parameters BASDAI and CRP (as opposed to the clear improvement of both parameters in responders) was observed during treatment resulting in a smaller decrease of the means for BASDAI, CRP and the other composite outcome parameter ASDAS (which contains also CRP) in the entire group (table 2, figure 2) compared with the binary outcome parameters ASAS40 and BASDAI50. It has to be investigated in a larger trial with ustekinumab whether such a dichotomy in the response, which has not been demonstrated in the trials with TNFα blockers for instance, can be confirmed or whether this was rather a chance finding in our small study.
There was a substantial (by 41% and 31% for the sacroiliac joints and for the spine, respectively) reduction of active inflammation as detected by MRI at week 24 in the entire group. This effect was again more prominent in patients with clinical response to ustekinumab in comparison with non-responders. In light of the relatively slow dynamic of the clinical response (figure 3), it might take longer than 24 weeks in order to observe the full effect of ustekinumab on active inflammation.
In this study, active inflammation in the sacroiliac joints on MRI was a clear predictor of clinical response (ie, ASAS40). Presence of active inflammation in the spine on MRI, elevated CRP, better function, younger age and shorter symptom duration demonstrated non-significant trends for the association with good response.
Data from a recent study in an animal model with some features resembling spondyloarthritis suggest that inhibition of IL-23 does suppress inflammation and inhibit osteoblasts through downregulation of IL-22.25 Thus, IL-23 might especially be an interesting treatment target in AS because long-term outcome is mostly defined by new bone formation and ankylosis while acute signs and symptoms are predominantly defined by inflammation.
The cellular source of IL-23 in AS is not well defined. We recently performed an immunohistochemical analysis of facet joints from AS patients and reported that IL-23 was predominantly expressed by myeloperoxidase-positive cells and, to a lesser extent, by macrophages.19 It is also not clear whether a potential effect of IL-23 blockade works through IL-17 inhibition or by other mechanisms, and whether the therapeutic effect we show here is solely mediated by IL-23 inhibition or alternatively or in addition through inhibition of IL-12. These open questions have to be addressed in subsequent investigations. Nonetheless, in the above-mentioned immunohistochemical study of subchondral bone marrow, IL-23 was clearly more strongly expressed than IL-12.19
In the current study, only AS patients were included who were not TNF failures in order to investigate whether ustekinumab might be a treatment option in AS. However, there is certainly also an unmet need for other treatments if TNFα blockers fail. Recent studies showed increased rates of response with ustekinumab compared with placebo in patients with Crohn's disease47 and psoriatic arthritis48 who failed previous TNFα blocker treatment. Thus, based on the data presented here, a similar study would be of interest in AS.
We chose a dose of 90 mg of ustekinumab per administration in this proof-of-concept study because this dose was slightly superior to 45 mg in studies for other indications such as psoriasis.28 Different dosages in the lower range should be compared with the 90 mg dose in future AS studies.
Recently, new classification criteria have been developed for patients with axial spondyloarthritis covering both patients with AS and patients in an earlier phase with the so-called non-radiographic axial spondyloarthritis.49 Such a new classification was necessary because the development of structural bony damage visible on x-rays, necessary to fulfil the modified New York criteria for AS, often takes years. Therefore, future studies with ustekinumab should also consider using the axial spondyloarthritis criteria for including patients with non-radiographic axial spondyloarthritis.
The clear limitations of this study are its open-label design and small sample size. However, we believe that even with these limitations of the pilot trial, an important signal of the possible therapeutic efficacy of ustekinumab in active AS was received.
In conclusion, this proof-of-concept trial with ustekinumab in active AS showed promising results and warrant confirmation in a placebo-controlled trial.
Acknowledgments
We are grateful to Beate Buß and Renate Lies for the study management; Esther Apt, Sebastian Leidig and Sabine Achtelstetter for the data management support; and Cristoph Thomann for the safety database management support. We further like to thank the following rheumatologists for the patient recruitment support: Henning Brandt, Hildrun Haibel, Sandra Hermann, In-Ho Song, Inge Spiller and Uta Syrbe.
References
Footnotes
Handling editor Tore K Kvien
-
Contributors All authors contributed to acquisition, analysis and interpretation of the data and drafting of the manuscript.
-
Funding The study was supported by an unrestricted research grant from Janssen-Cilag.
-
Competing interests DP: lecture/speakers honoraria from Abbvie, Merck Sharp & Dohme, Pfizer and UCB. KGAH: lecture/speakers honoraria from Abbvie, Janssen Research & Development, MSD. JS: research grants from Merck Sharp & Dohme, Pfizer, Abbvie and Janssen-Cilag; consulting fees from F. Hoffmann-La Roche, Eli Lilly and Company, Merck Sharp & Dohme, Pfizer, Abbvie and Novartis; expert testimony fees from Abbvie and lecture/speakers honoraria from Abbvie, Merck Sharp & Dohme, Pfizer and UCB.
-
Ethics approval The study protocol was approved by the ethics committee of the federal state Berlin, Germany. Written informed consent was obtained from all patients.
-
Provenance and peer review Not commissioned; externally peer reviewed.