Article Text

Abstract
Objective Alterations in DNA methylation patterns have been found to correlate with several diseases including osteoarthritis (OA). The aim of this study was to identify, for the first time, the genome-wide DNA methylation profiles of human articular chondrocytes from OA cartilage and healthy control cartilage samples.
Methods DNA methylation profiling was performed using Illumina Infinium HumanMethylation27 in 25 patients with OA and 20 healthy controls. Subsequent validation was performed by genome-wide expression analysis using the Affymetrix Human Gene 1.1 ST array in an independent cohort of 24 patients with OA. Finally, the most consistent genes in both assays were amplified by quantitative reverse transcriptase PCR in a validation cohort of 48 patients using microfluidic real-time quantitative PCR. Appropriate bioinformatics analyses were carried out using R bioconductor software packages and qBase plus software from Biogazelle.
Results We found 91 differentially methylated (DM) probes, which permitted us to separate patients with OA from healthy controls. Among the patients with OA, we detected 1357 DM probes that identified a tight cluster of seven patients who were different from the rest. This cluster was also identified by genome-wide expression in which 450 genes were differentially expressed. Further validation of the most consistent genes in an independent cohort of patients with OA permitted us to identify this cluster, which was characterised by increased inflammatory processes.
Conclusions We were able to identify a tight subgroup of patients with OA, characterised by an increased inflammatory response that could be regulated by epigenetics. The identification and isolation of this subgroup may be critical for the development of effective treatment and disease prevention.
- Osteoarthritis
- Knee Osteoarthritis
- Chondrocytes
Statistics from Altmetric.com
Introduction
Osteoarthritis (OA) is a chronic musculoskeletal disease related to aging that is characterised by the progressive destruction of the extracellular matrix of articular cartilage, as well as bone remodelling and synovial inflammation, which leads to pain and severe impairment of mobility. It is the main cause of work incapacity and one of the most common reasons for visiting a primary care physician; its prevalence reaches 40% of people over the age of 70,1 and there is no basic treatment. Age, obesity, body mass index, behavioural influences and both nuclear and mitochondrial genetics are all risk factors2–4; however, in the last few years, the role played by epigenetics in the pathogenesis of the disease has been investigated in more detail.5–8
Epigenetics is defined as heritable changes in genome function without changes in the DNA sequence. While the genetic code is the same for every somatic cell, epigenetic changes are usually confined to specific cells or tissues, or even to specific cells within a tissue. Epigenetics mechanisms must be considered in order to understand the gene–environment interactions that lead to complex diseases,9 and OA is no exception. DNA methylation is the best characterised epigenetic mechanism, and its major goal is gene silencing.10 It consists of the addition of a methyl group (CH3) to cytosine that is 5′ to guanine to form methylated cytosine (MeC). These sites are usually located in promoter regions and are called CpG sites.
DNA methylation is an essential mechanism for (i) imprinting of specific genes, (ii) X chromosome inactivation in women, and (iii), important in the present context, cell-type-specific gene expression. In this regard, since it has been suggested that osteoarthritic chondrocytes possess a modulated phenotype11 ,12 by which several genes are up- or down-regulated compared with normal chondrocytes,13 and the regulatory mechanisms responsible for these alterations have not yet been clarified, it has been suggested that epigenetic changes in the methylation status of the promoter region of the genomic DNA contribute to the pathology of OA.5 ,12
To date, epigenetic gene regulation has been widely studied in cancer,14–16 but very few studies on OA have been performed. One of the first investigations of OA showed that overexpression of cartilage-degrading enzymes by late-stage OA chondrocytes, mainly matrix metalloproteinase-3 (MMP-3), MMP-9, MMP-13 and ADAMTS-4, may have resulted from epigenetic changes in the methylation status of CpG sites in the promoter regions of these enzymes12 ,17; in addition, epigenetic changes in the promoter region of the leptin gene result in modulated expression of MMP-13.13 It has also been postulated that a DNA demethylation process takes place at specific CpG sites in the interleukin-1β promoter in response to inflammatory cytokines in human chondrocytes,18 and even the DNA methylation pattern in the promoter region of this gene is also altered after treatment with glucosamine and a nuclear factor-kappa β inhibitor.19 It has furthermore been demonstrated that the genetic effect of the rs143383 single-nucleotide polymorphism on expression of growth differentiation factor 5 (GDF5) in OA disease is regulated epigenetically by DNA methylation.20 Other genes related to the oxidative stress process, such as manganese superoxide dismutase, which is repressed in OA chondrocytes,21 has been shown to be regulated by epigenetics.22 In contrast, a total DNA methylation assay performed by Sesselmann and colleagues23 showed no differences between healthy control and OA cartilage by chromatographic approaches. Another recent study, consisting of a genome-wide methylation approach, revealed differentially methylated (DM) regions in osteoporotic and OA bones.24
Taking all this into account, we examined for the first time the genome-wide DNA methylation profile of human articular chondrocytes from OA and healthy control cartilage samples to identify profiles of DNA methylation in OA disease. By examining the gene pathways involved, as well as the genomic context of the loci with OA-associated methylation, we provide insight into potentially altered biological processes related to these profiles.
Materials and methods
The scheme we followed to carry out the present work is described in online supplementary figure S1.
Samples
Three groups of samples from Hospital Universitario A Coruña were analysed (see online supplementary table S1). All patients included in this study had knee OA K/L grade IV and underwent knee prosthesis surgery; normal human cartilages were obtained at autopsy from cadavers with no macroscopic signs of OA. All cartilage samples were collected from the central area of the tibial plateau of the knee and represent a mixture of superficial, intermediate and deep layers of articular cartilage. Informed consent was obtained from all participants, and the study was approved by the clinical ethics committee of Galicia (see online supplementary materials and methods).
Nucleic acid (DNA and RNA) isolation and reverse transcription
DNA was isolated following a home-made CetylTrimethyl Ammonium Bromide (CTAB) and chloroform-based method after breaking up the cartilage. For RNA isolation, cartilage samples were disrupted using a mortar, after freezing in liquid nitrogen before using the RNeasy Mini Kit (Qiagen, Duesseldorf, Germany). After isolation, the RNA was reverse transcribed to cDNA (see online supplementary materials and methods).
DNA methylation profiling
DNA methylation profiling was performed using the Infinium HumanMethylation27 beadchip (Illumina, San Diego, California, USA), which allows interrogation of 27 578 highly informative CpG sites located within the proximal promoter regions of 14 495 genes and 110 microRNAs (see online supplementary materials and methods). The methylation data have been deposited in the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) with the GEO accession number GSE43269.
Data filtering, normalisation and analysis of methylation data
As a result of data filtering, four samples (two patients with OA and two healthy controls) showed a detection p value >0.05 in more than 25% of all probes; consequently, these samples were removed from further analyses, resulting in 23 patients with OA and 18 healthy controls. After normalisation, M-values followed a bimodal distribution (see online supplementary figure S2). The statistical procedures used to analyse the DM sites are described in online supplementary materials and methods.
Unsupervised clustering of methylation data
A resampling-based unsupervised consensus clustering of the DNA methylation dataset, using the 23 837 selected probes that followed the initially established criteria, was performed to distinguish different groups25 (see online supplementary materials and methods).
Gene expression profiling and data analysis from gene expression
The platform used for this purpose was the Affymetrix Human Gene 1.1 ST array plate (Affymetrix, Santa Clara, California, USA). A principal component analysis (PCA) using the 7742 probes selected after initial non-specific filtering was carried out in order to visualise possible clusters within samples. The normalisation processes as well as the procedure to analyse the differentially expressed probes are described in online supplementary materials and methods. The expression data have been deposited in the NCBI GEO (http://www.ncbi.nlm.nih.gov/geo/) with the GEO accession number GSE43191.
Gene ontology analyses
Functional characteristics consisted of an over-representation analysis of the gene ontology (GO) categories which appeared enriched in the selected lists of the DM/expressed genes examined using the DAVID (Database for Annotation, Visualisation and Integrated Discovery) bioinformatics database functional tool.26
Validation of genes
The expression levels of those genes in which an inverse relationship between methylation and expression was detected were subsequently analysed in an independent cohort of 48 patients with OA using microfluidic real-time quantitative PCR. The data analyses were carried out with qBase plus software (Biogazelle, Ghent University, Belgium) and R Bioconductor software, and included qualitative, quantitative and hierarchical approaches to validate and identify clusters of patients. The genes hypoxanthine phosphoribosyltransferase-1 (HPRT1) and ribosomal protein L13A (RPL13A) were used as constitutive genes. Four of the 48 samples (RNAs) did not meet the quality criteria and were therefore removed from further analyses.
Results
DNA methylation pattern in patients with OA and healthy controls
A total of 91 DM probes in patients with OA and healthy controls were detected (table 1), permitting us to distinguish the two groups of samples (see online supplementary figure S3a). Of these, 37 were less methylated in OA samples and 54 were more methylated in OA samples compared with healthy controls. The runt-related transcription factor-1 (RUNX1) gene was the least methylated gene and the msh homeobox-1 (MSX1) gene was the most methylated gene in patients with OA compared with healthy controls.
Differentially methylated probes in osteoarthritic cartilage samples and healthy cartilage samples sorted by decreased mean difference in β values on each of the main categories
The biological process associated with the less methylated genes in patients with OA were those related to the inflammatory/defence response (p=0.019), as well as those related to the positive regulation of transcription activity (p=0.011); on the other hand, the altered processes related to genes with increased methylation levels in patients with OA were those involved in the regulation of phosphorylation (p=0.006) and mitogen-activated protein kinase activity (p=0.004).
The unsupervised clustering resulting from the 23 837 selected probes showed three different clusters of samples (see online supplementary figure S3b): cluster 1 consisted of 14 patients with OA and six healthy controls, cluster 2 consisted of two patients with OA and 11 healthy controls, and cluster 3 was formed by seven patients with OA and one healthy control. This technique showed cluster 3 to be a particularly tight cluster of patients with a characteristic DNA methylation profile.
We again performed unsupervised clustering with the selected probes including only the 23 patients with OA. The results showed the same cluster of patients as cluster 3 above, which could be distinguished from the rest of the patients (see online supplementary figure S3c). We therefore based our subsequent analyses on cluster membership from the above clustering method that identified a different subgroup of OA.
Identification and characterisation of OA subgroups
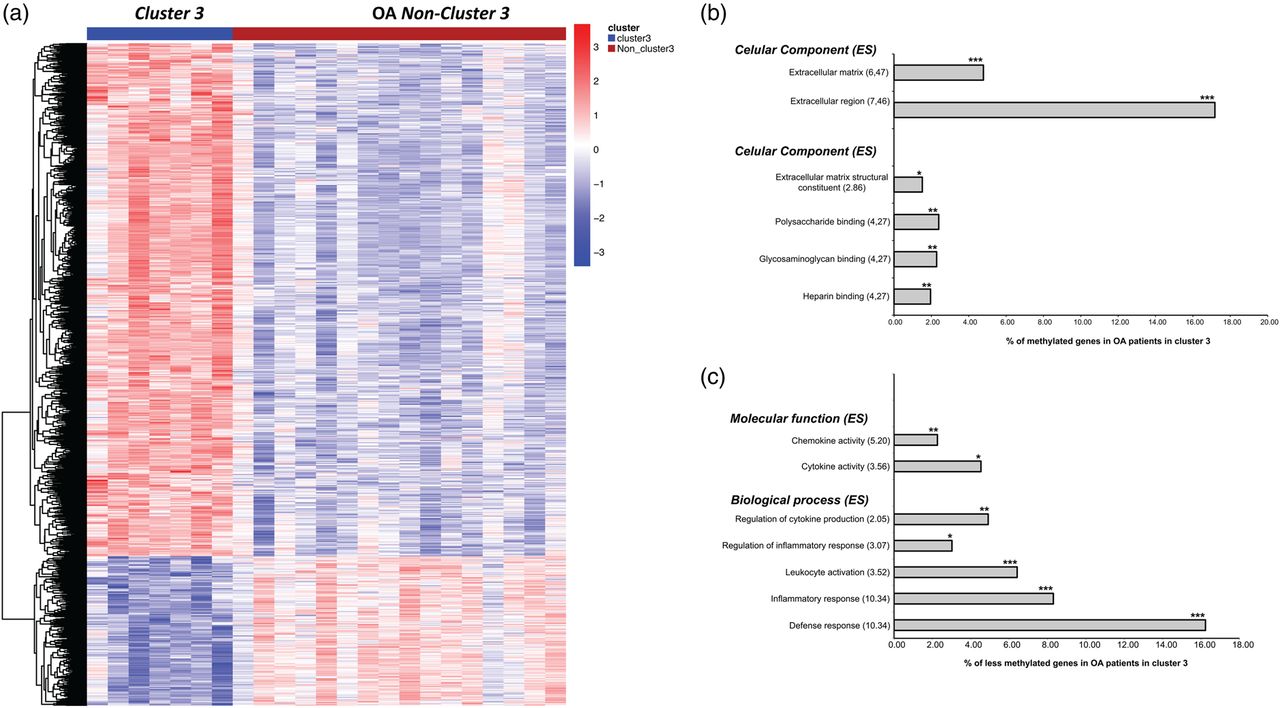
When we compared the DM probes of patients in cluster 3 with the rest of the patients, we identified a total of 1357 significant probes (see online supplementary table S2) that separated the two subgroups of patients (figure 1A). Of these, 306 were less methylated in samples from cluster 3, and 1051 appeared more methylated in this group of samples. Of the less methylated genes in cluster 3, RUNX1, RUNX2 and genes related to inflammatory response were notable, whereas some genes related to the synthesis of collagens, such as collagen XV α-1 (COL15A1), collagen XVI α-1 (COL16A1), collagen III α-1 (COL3A1), collagen IX α-1 (COL9A1), collagen VI α-3 (COL6A3), procollagen C-endopeptidase enhancer (PCOLCE), collagen XVIII α-3 (COL18A1), collagen XI α-1 (COL11A1) and collagen XXI α-1 (COL21A1), and other cartilage and extracellular matrix-related genes, such as cartilage intermediate layer protein, GDF5, extracellular matrix-2 (ECM2) and chondroitin sulfate proteoglycan-4 (CSPG4), showed significantly increased methylation in this subgroup of patients.

Hierarchical analysis and potentially altered biological process gene ontology (GO) of the differentially methylated probes in patients with osteoarthritis (OA) in cluster 3 and non-cluster 3 patients. (A) Heatmap showing the 1357 differentially methylated probes in patients with OA in cluster 3 and non-cluster 3 patients; (B) GO analysis comparing the methylated genes in patients with OA in cluster 3 and non-cluster 3 patients; (C) GO analysis comparing the less methylated genes in patients with OA in cluster 3 and non-cluster 3 patients. ES, enrichment score. ***p≤0.0001, **p≤0.01, *p≤0.05 with false discovery rate correction for multiple testing.
Most of the methylated genes in patients in cluster 3 corresponded to the extracellular region (p<0.0001), including the extracellular matrix (p<0.0001); the altered molecular functions were those related to extracellular matrix structural constituent (p=0.05), polysaccharide binding (p=0.008), glycosaminoglycan binding (p=0.009) and heparin binding (p=0.011) (figure 1B). With regard to the less methylated genes (figure 1C), the altered molecular functions were those related to chemokine activity (p=0.005) and cytokine activity (p=0.018), and the biological processes were those related to the regulation of cytokine production (p=0.002), regulation of inflammatory response (p=0.014), leucocyte activation (p<0.0001), inflammatory response (p<0.0001) and defence response (p<0.0001).
Identification of the subgroups of OA by genome-wide expression analysis
To identify the subgroup of patients with OA revealed by the genome-wide DNA methylation approach, we performed a genome-wide expression assay in an independent cohort of 24 OA samples. In the first step, we analysed the microarray data obtained (23 OA samples and 7742 probes retained after array normalisation and non-specific filtering) using PCA as the unsupervised clustering method. This approach separated the OA population into two subgroups: a highly homogeneous group (named cluster A) and a heterogeneous group of six patients (named cluster B) (figure 2).

Principal component analysis (PCA) of osteoarthritis (OA) samples using the 7742 probes selected from expression data after initial non-specific filtering. The PCA clearly shows the distribution of the two OA subgroups (cluster A and cluster B).
Analysis of differentially expressed genes in the two subgroups of patients with OA
We created two groups of OA samples according to the clusters identified by the PCA method. When patients were grouped according to this distribution (cluster A versus cluster B), a total of 450 probes were found to be differentially expressed between the two groups (see online supplementary table S3). Of these, 262 were upregulated in patients in cluster B, and 188 were downregulated in this cluster. Heatmap representation showed the differentiation between the two groups of patients (figure 3A).

Hierarchical analysis and potentially altered biological process gene ontology (GO) of the differentially expressed probes in patients with osteoarthritis (OA) in cluster B and non-cluster B patients. (A) Heatmap showing the 450 differentially expressed probes in patients in cluster B and non-cluster B patients; (B) GO analysis comparing the upregulated genes in patients in cluster B with non-cluster B patients. ES, enrichment score. ***p≤0.0001, **p≤0.01, *p≤0.05 with false discovery rate correction for multiple testing.
Most of the upregulated genes in patients in cluster B belonged to the plasma membrane (p<0.0001), and the biological processes related to the less methylated genes in the patients in cluster 3, such as defence response (p<0.0001), inflammatory response (p<0.0001), leucocyte activation (p<0.0001), regulation of cytokine production (p=0.0045) and chemokine activity (p=0.0028), were significantly over-represented in this group of patients also. In addition, increased representation of superoxide anion generation (p=0.0206) and increased synthesis of unsaturated fatty acids (p=0.0349) were detected (figure 3B).
Comparative analysis and validation of the results by quantitative reverse transcriptase (qRT)-PCR
The expression pattern detected in patients in cluster B seems to indicate that this group of patients is closely related to those identified according to the methylation pattern in patients in cluster 3, showing this subgroup of patients to be a tight group characterised by an increased inflammatory response which could be modulated epigenetically leading to reduced synthesis of extracellular matrix constituents (figures 1 and 3).
With regard to the above, we identified 47 significantly altered genes that showed an inverse relationship between the two approaches (table 2). Of these, 15 were methylated in patients in cluster 3 and also downregulated in patients in cluster B; meanwhile, 32 were less methylated in patients in cluster 3 and also upregulated in patients in cluster B, representing 10.4% (47/450) correlated genes.
List of the 47 significantly altered genes that showed an inverse relationship between methylation and expression in both methylation and expression cohorts
We selected for validation, in an independent cohort of 48 patients, a total of eight (less methylated and upregulated) genes concentrating on their increased B-statistic value in both assays: lysosomal protein transmembrane-5 (LAPTM5), T-cell specific surface glycoprotein CD28 (CD28), Fc fragment of IgG low affinity IIb receptor CD32 (FCGR2B), integrin alpha M (ITGAM), pleckstrin (PLEK), lymphocyte cytosolic protein 2 (LCP2), guanine nucleotide-binding protein (G protein) α15 (GNA15) and kynureninase (KYNU). We also included lymphocyte antigen 96 (LY96) for analysis because of its high expression and low methylation levels in some patients in cluster B and cluster 3, respectively.
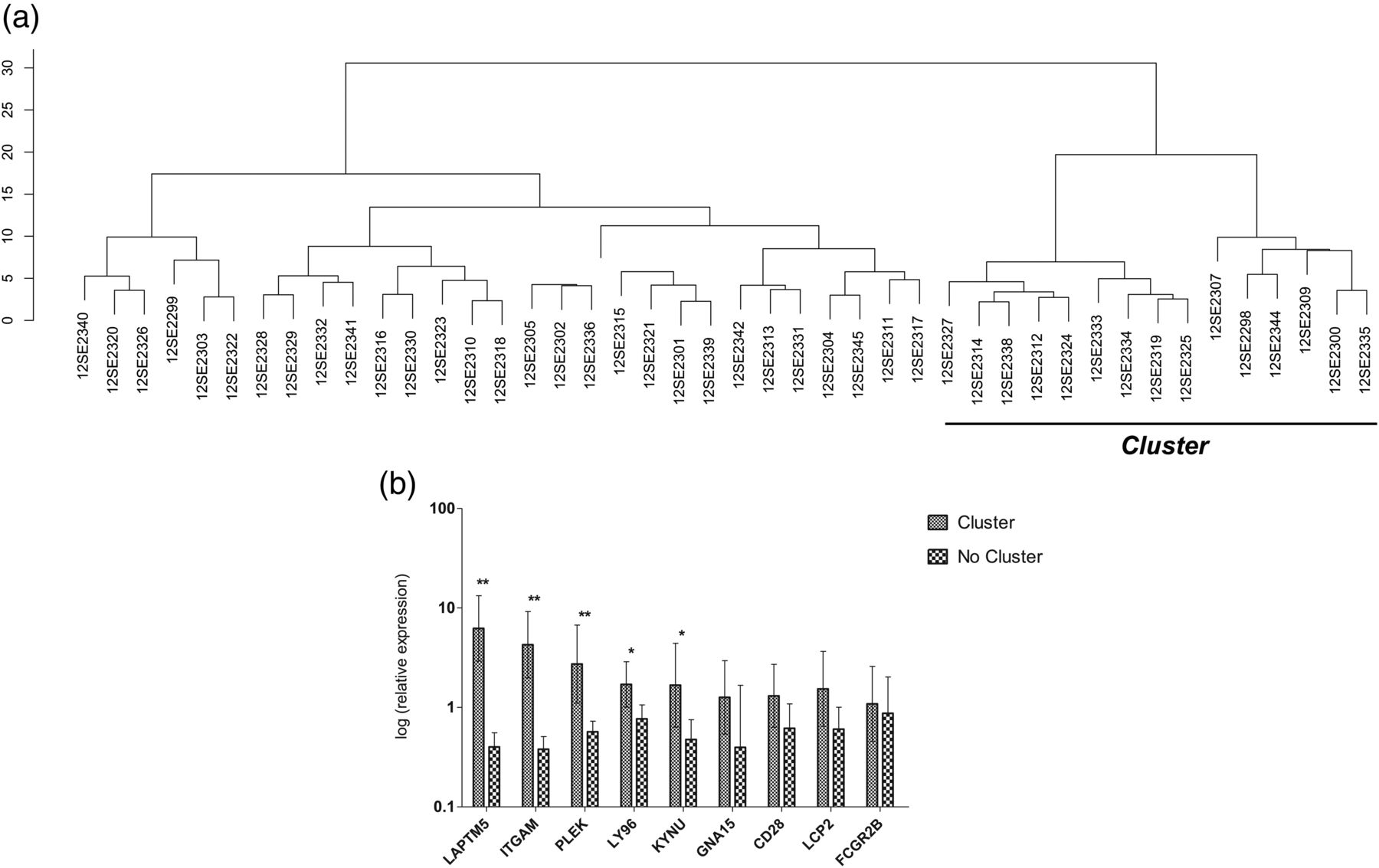
As expected, not all the samples amplified all the selected genes, with only six of the 44 samples showing amplification for all genes (see online supplementary table S4). On the basis of the expression of these genes in the 44 samples, the hierarchical clustering performed revealed the existence of a cluster of 15 patients, who were different from the remaining 29 (figure 4A), as observed in both methylation (cluster 3) and expression (cluster B) assays. The six samples that amplified the nine selected genes were also included in this cluster. A comparison of the expression levels of the selected genes in these 15 patients and the remaining 29 showed significant differences between the two subgroups, with most of the genes being upregulated in the first subgroup (figure 4B). The most significant differences in gene expression between the two groups corresponded to LAPTM5, ITGAM and PLEK genes, but LY96 and KYNU also showed significantly increased expression.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Hierarchical clustering and mean expression levels in the validation cohort of osteoarthritis (OA) samples. (A) Cluster of patients obtained by concentrating on the expression levels of the nine selected genes. (B) Mean expression levels of the nine selected genes in both clusters. Vertical bars indicate 95% CI. **p≤0.0001, *p≤0.05 after Mann–Whitney non-parametric test.
Discussion
We analysed, for the first time in articular cartilage, the genome-wide DNA methylation profiles of a cohort of 25 patients with OA and 20 healthy controls. Furthermore, because of the subgroup of patients detected by this assay, we tried to identify this cluster of patients by a genome-wide expression approach in a population of 24 patients with OA. Finally, we validated the most consistent common genes resulting from the above assays by qRT-PCR in an independent cohort of 48 patients with OA.
The comparison of the methylation patterns of patients with OA and healthy controls showed 91 DM probes, with RUNX1 being the most DM and least methylated gene in OA. The RUNX1 gene is a critical transcription factor during embryogenesis which is thought to be involved in the development of normal haematopoiesis, neoplastic disease27 and chondrogenesis28 and has also been related to cell death,29 which is also a characteristic of OA.28 ,30–32 Contradictory results with regard to the expression of this gene in OA have been described.33 ,34 In contrast, MSX1 was the most methylated gene in OA; this gene is a member of the MSX family, is associated with bone formation, and has been found to be increased in patients with OA compared with healthy controls in a genome-wide expression study.33 As none of these genes have previously been shown to be epigenetically modulated in OA, they can be considered to be novel DM loci in this pathology.
In summary, the biological processes associated with the whole set of 91 DM probes in patients with OA and healthy controls are mainly related to the inflammatory/defence response (demethylated), indicating not only that inflammation is a key process in OA,35 but also that it may be regulated by epigenetics.
An important finding of this work is the existence of two OA subgroups (clusters) as a result of the unsupervised clustering carried out in the whole samples. This approach is usually used in methylation/expression studies when it is suspected that different subsets of clinical samples might occur.36–38 On this basis, samples belonging to cluster 3 are characterised by decreased synthesis of extracellular matrix constituents and increased inflammatory response. Interestingly, in addition to a wide range of types of collagens and extracellular matrix-related genes, in this group of patients we found a greater degree of methylation in the GDF5 gene, as described previously,20 as well as a marked decrease in methylation of the RUNX2 gene. The RUNX2 gene is a transcription factor involved in osteoblastic differentiation and skeletal morphogenesis, and increased expression has been found in OA cartilage.35 ,39
Previous studies have shown epigenetic (demethylation) regulation of some of the cartilage-degrading enzymes, such as MMP-3, MMP-9, MMP-13 and ADAMTS-4.12 ,17 However, in the present work we did not find any significant difference between patients with OA and healthy controls, and only a small increase in methylation was detected for both MMP-3 and ADAMTS-14 in patients in cluster 3 compared with non-cluster 3 patients. In order to explain this, it should be stated that: (i) the overall methylation levels of a particular gene may be important in regulating this gene rather than the methylation status of specific CpG dinucleotides,20 whereby it is possible that the CpG loci analysed by the above authors are not included in the Infinium HumanMethylation27 beadchip; (ii) these cartilage-degrading enzymes are mainly (modestly) expressed by chondrocytes in the superficial zones of the OA cartilage,40 ,41 and, as we extracted the DNA from the whole (normal and OA) cartilage, this discrepancy would not be unexpected; (iii) the data related to mRNA expression for MMP-3 found in the literature are very contradictory12; and (iv) despite global DNA methylation levels decreasing with age,42–44 a reduction in DNA methylation is technically more difficult to detect.16
To identify this tight cluster of patients and validate the results of the methylation assay, we performed a genome-wide expression analysis in an independent cohort of patients, which also revealed the existence of a tight cluster of patients (cluster B). Moreover, a large coincidence was detected between the most significant biological processes related to the less methylated genes in the patients in cluster 3 and the most significant biological processes related to the upregulated genes in cluster B. Therefore, an increased number of genes related to cytokine production and inflammatory/defence response were significantly altered in this subgroup of patients. With regard to this, this subgroup of patients was also characterised by an increased inflammatory response. As the group of patients that comprise cluster B are characterised by increased body mass index (personal data), this may be related to systemic factors induced by obesity (eg, adipokines, increased synthesis of fatty acids) and/or the increased oxidative stress detected in this subgroup of patients (see figure 3B), as previously proposed.45 ,46
Despite the use of three independent groups of samples for each of the approaches performed in this work, a marked and common finding is the identification of a tight subgroup of patients with all of the assays performed. We can therefore rule out the possibility that this cluster is based on features other than the pathogenesis of the disease itself, such as age or gender. All the models were adjusted for age, and the mean age for the samples that comprise the different clusters is very similar; moreover, despite the higher frequency of women in all groups, no difference in gender distribution was detected between clusters (see online supplementary table S1).
In addition, an inverse relationship between methylation and expression was detected for 47 genes (10.4%), and the validation by qRT-PCR of some of the genes showing the most consistent relationship was able to identify this tight cluster of patients, indicating not only significant differences between the two groups, but also candidate genes that could be used as new therapeutic markers for OA such as PLEK, LAPTM5, ITGAM, Y96 and KYNU.
In summary, as OA can be considered the final common pathway of joint destruction for multiple different pathophysiological aetiologies which define the existence of different phenotypes of OA,47 the identification and isolation of these phenotypes (or subgroups) may be critical for the development of effective treatment and disease prevention.47 In this regard, this work shows not only that patients and healthy controls have different methylation patterns, but also the existence of a tight cluster of patients, corroborated by a genome-wide expression assay. Both approaches reveal a higher-than-usual inflammatory response significantly represented in this subgroup of patients; this condition may be caused by increased oxidative stress and fatty acid synthesis, leading to decreased synthesis of extracellular matrix constituents. By using some of the genes with a consistent correlation between methylation and expression, we were able to identify novel genes in a subgroup of patients that may emerge as new therapeutic candidate markers of OA disease. However, as methylation is a dynamic process prone to changes in the environment (disease) and as the age-related loss of normal epigenetic patterns is a possible mechanism for the late onset of common human diseases (such as OA), further studies of, for example, dimethyltransferases need to be performed to ascertain if changes in DNA methylation patterns play a major role in OA pathogenesis or, on the contrary, these changes are a result of altered OA chondrocytes or even a combination of both scenarios.
Acknowledgments
We would like to express our appreciation of Lourdes Sanjurjo and Maria Dolores Veloand of the Department of Orthopaedic Surgery of CHU A Coruna. This study was supported by grants from Fundacion Espanola de Reumatologia (programa GEN-SER) and from Fondo Investigacion Sanitaria (CIBERCB06/01/0040)-Spain, Fondo Investigacion Sanitaria-PI 08/2028, Ministerio Ciencia e Innovacion PLE2009-0144, with a contribution of funds from FEDER (European Community). IR-P was supported by Contrato de Apoyo a la Investigacion-Fondo Investigacion Sanitaria (CA10/01564). MEV-M was supported by a grant from Programa Rio Hortega (Fondo de Investigacion Sanitaria). Methylation assay services were provided by the Spanish ‘Centro Nacional de Genotipado (CEGEN-ISCIII)’ (http://www.cegen.org).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online figures
- Data supplement 2 - Online methods
- Data supplement 3 - Online table 1
- Data supplement 4 - Online table 2
- Data supplement 5 - Online table 3
- Data supplement 6 - Online table 4
Footnotes
Handling editor Tore K Kvien
-
Contributors JF-T performed data filtering, normalisation and analysis of methylation data. He also participated in gene expression profiling and gene expression data analysis, as well as the gene ontology analyses. He helped to draft the manuscript and has given final approval of the version to be published. EC-P and MF-M were involved in nucleic acid (DNA and RNA) isolation and reverse transcription. JLF and AM carried out DNA methylation profiling. AS-H and MEV-M carried out gene validation. NO and CF-L collected the samples and checked clinical histories for the inclusion and exclusion criteria. Both collected and analysed the radiographic information of the samples. IR-P was involved in the conception and design of the study; he helped to draft the manuscript and has given final approval of the version to be published. He was also involved in the bioinformatics analysis of the results. FJB conceived the study, and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval Clinical ethics committee of Galicia-Spain.
-
Provenance and peer review Not commissioned; externally peer reviewed.