Article Text

Abstract
Objective To evaluate the efficacy and safety of secukinumab, a fully human, anti-interleukin (IL)-17A monoclonal antibody, in patients with psoriatic arthritis (PsA).
Methods 42 patients with active PsA fulfilling ClASsification for Psoriatic ARthritis (CASPAR) criteria were randomly assigned (2:1) to receive two intravenous secukinumab doses (10 mg/kg; n=28) or placebo (n=14) 3 weeks apart. The primary endpoint was the proportion of American College of Rheumatology (ACR) 20 responses at week 6 for secukinumab versus placebo (one-sided p<0.1).
Results Primary endpoint: ACR20 responses at week 6 were 39% (9/23) for secukinumab versus 23% (3/13) for placebo (p=0.27). ACR20 responses were greater with secukinumab versus placebo at week 12 (39% (9/23) vs 15% (2/13), p=0.13) and week 24 (43% (10/23) vs 18% (2/11), p= 0.14). At week 6, ‘good’ European League Against Rheumatism response was seen in 21.7% (5/23) secukinumab versus 9.1% (1/11) placebo patients. Compared with placebo at week 6, significant reductions were observed among secukinumab recipients for C reactive protein (p=0.039), erythrocyte sedimentation rate (p=0.038), Health Assessment Questionnaire Disability Index (p=0.002) and Short Form Health Survey (SF-36; p=0.030) scores. The overall adverse event (AE) frequency was comparable between secukinumab (26 (93%)) and placebo (11 (79%)) recipients. Six serious AEs (SAEs) were reported in four secukinumab patients and one SAE in one placebo patient.
Conclusions Although the primary endpoint was not met, clinical responses, acute-phase reactant and quality of life improvements were greater with secukinumab versus placebo, suggesting some clinical benefit. Secukinumab exhibited satisfactory safety. Larger clinical trials of secukinumab in PsA are warranted.
- Psoriatic Arthritis
- Cytokines
- TNF-alpha
Statistics from Altmetric.com
Introduction
Psoriatic arthritis (PsA) is a chronic inflammatory joint disorder associated with synovitis, enthesitis and, in the majority of patients, classical psoriatic skin lesions. It affects 0.3%–1% of the general population and up to 30% of patients with psoriasis.1 ,2 The natural course of PsA involves significant disability and reduced life expectancy.3 ,4 The primary goals of treatment are to arrest disease progression, to improve quality of life and ultimately to achieve clinical remission.5 Therapeutic interventions for PsA include conventional disease-modifying antirheumatic drugs (DMARDs) and biological agents that target the proinflammatory cytokine tumour necrosis factor α (TNFα).6 ,7 Rigorous use of these agents has improved the management of PsA in recent years; however, significant unmet clinical needs remain. These include limited drug tolerance; non-responsiveness, partial responsiveness or development of therapeutic resistance to existing agents; and, in some patients, lack of prevention of long-term structural damage.
Synovial tissue from PsA patients is characterised by increased levels of interleukin (IL)-17A produced by T cells and possibly by non-T cells.8–10 In PsA, IL-17A is functionally active, often operating in synergy with adjacent inflammatory cytokines, and it regulates the expression of cytokines and matrix metalloproteinases, which promote pathogenesis.11 An IL-17-inducible gene signature identified in keratinocytes from patients with psoriasis vulgaris skin lesions includes chemoattractants, such as IL-8 and chemokine (C–C motif) ligand 20 (CCL20), and innate molecules, such as S100 calcium-binding proteins A8 and A9 (S100A8/A9) and human β-defensin-2 (hBD-2; also known as β-defensin-4).12 Serum hBD-2 has recently been suggested to be a useful surrogate marker for disease activity in psoriasis.13 Moreover, lesional hBD-2 mRNA has been found to be significantly downregulated upon treatment with etanercept14 and secukinumab.15
Secukinumab is a fully human, high-affinity, anti-IL-17A monoclonal antibody that binds to and neutralises IL-17A. In proof-of-concept and phase II trials, secukinumab showed therapeutic potential in rheumatoid arthritis, moderate-to-severe plaque psoriasis, non-infectious uveitis and ankylosing spondylitis.15–18 In the present study, we evaluated the efficacy and safety of secukinumab in patients with active, moderate-to-severe PsA. In addition, we sought to explore the relationship between serum hBD-2 and clinical responses to secukinumab in this patient population.
Materials and methods
Study design and patient population
This was a randomised, double-blind, placebo-controlled, multicentre, 24-week, phase IIa study of secukinumab (two intravenous doses of 10 mg/kg given 21 days apart) for the treatment of active PsA. The study was conducted from 18 March 2009 to 22 December 2010, at 11 centres in three countries: Germany, The Netherlands and the UK.
Male and female patients aged 18–65 years fulfilling the following criteria were eligible for enrolment: moderate-to-severe PsA diagnosed according to the ClASsification for Psoriatic ARthritis (CASPAR) criteria19 and involvement of three or more swollen and tender peripheral joints; Physician's Global Assessment score ≥40, based on a visual analogue scale (VAS 0–100 mm); inflammatory pain score ≥40 (VAS 0–100 mm); disease inadequately controlled on ≥1 DMARD given for ≥3 months at the maximum tolerated dose; and a rheumatoid factor level ≤100 IU with a negative cyclic citrullinated protein ELISA result. Key exclusion criteria included the presence of arthritis fulfilling the classification criteria for rheumatoid arthritis or a seronegative spondyloarthropathy fulfilling the modified New York classification criteria for ankylosing spondylitis; history or presence of malignancy; positive blood test result for hepatitis B, hepatitis C or HIV infection; active systemic infection within 2 weeks of screening; severe, progressive or uncontrolled concomitant diseases; kidney impairment; or severe cardiac disease. Considerations regarding concomitant medications are summarised in the online supplementary appendix.
Informed consent was obtained from each patient before randomisation. The study was conducted according to the ethical principles of the Declaration of Helsinki and approved by the Independent Ethics Committee or Institutional Review Board for each centre. The study is registered with ClinicalTrials.gov (#NCT00809614), and the protocol is available from the sponsor.
Study procedures and evaluations
Forty-two patients were randomly assigned (2:1) to receive two doses of either secukinumab 10 mg/kg (n=28) or placebo (n=14). Secukinumab or placebo was administered intravenously at baseline (day 1) and on day 22. Patients returned on day 2 to provide a 24-hour post-infusion blood sample for pharmacokinetic and biomarker (mRNA and cytokine) analyses, then weekly (days 8 and 15) for response assessments and safety evaluations. On day 22, patients received the second infusion of secukinumab 10 mg/kg or placebo and then were observed for up to 21 weeks.
Novartis generated the randomisation list using a validated system. A quality assurance group reviewed the randomisation list, which was locked following approval. Patients and investigators were blinded to the treatment allocation, and the appearance of the placebo was identical to that of secukinumab to maintain blinding.
Disease assessments (tender and swollen joints) were made according to American College of Rheumatology (ACR) response criteria,20 Psoriatic Arthritis Response Criteria (PsARC),21 with distal interphalangeal joints included in the joint count, and the Disease Activity Score 28 (DAS28).22 The investigator or a designated qualified individual performed the joint counts. Joint tenderness and swelling were graded present (1) or absent (0). Patient and physician global assessments of disease activity were recorded on a 0–100 mm VAS, and a similar scale was used to assess pain intensity (VAS 0–100 mm). Other assessments included the European League Against Rheumatism (EULAR) response criteria, defined using the DAS28 (good responders were patients with an improvement in DAS28 from baseline of >1.2 and a present score of ≤3.2; moderate responders had an improvement of >0.6 to ≤1.2 and a present score of >3.2 and ≤5.1; non-responders had an improvement of ≤0.6 and a present score of >5.1);23 the Psoriasis Area and Severity Index (PASI 0 (no disease) to 72 (maximal disease)),24 which assessed the extent of psoriasis on four body surface areas (head, trunk, upper limbs and lower limbs) and the degree of plaque erythema, scaling and thickness; the Maastricht Ankylosing Spondylitis Enthesitis Score (MASES)25 ,26 and the Spondyloarthritis Research Consortium of Canada (SPARCC) score,27 which evaluated 13 and 18 enthesis sites, respectively; and the Leeds Dactylitis Index (LDI) basic score.28 The LDI basic score was derived by measuring the ratio of the circumference of the affected digit to the circumference of the digit on the opposite hand or foot, using a minimum difference of 10% to define a dactylitic digit; the ratio of the circumference was multiplied by a tenderness score (1 for tender, 0 for non-tender). C reactive protein (CRP) levels were measured with a high-sensitivity assay. Erythrocyte sedimentation rate (ESR) was measured locally using a standard kit supplied by the central lab. Patients’ health-related quality of life (HRQoL) was assessed using two validated questionnaires: the Short Form Health Survey (SF-36)29 and the Health Assessment Questionnaire Standard Disability Index (HAQ-DI).30
Study endpoints
The primary endpoint was the proportion of patients achieving an ACR20 response at week 6, defined by the following three conditions: ≥20% improvement in the number of tender joints (based on 68 joints), ≥20% improvement in the number of swollen joints (based on 66 joints) and ≥20% improvement in three of five additional domains (Patient's Global Assessment, Physician's Global Assessment, pain, disability measured by the HAQ and acute-phase reactant measured by CRP). The result was considered statistically significant if the one-sided p value was <0.1.
Key secondary endpoints were safety and tolerability at weeks 1, 2, 6, 8, 12, 16 and 24; the proportion of patients achieving ACR20, ACR50 and ACR70 responses (with the latter two defined in a manner similar to the ACR20 response but requiring improvements of ≥50% and ≥70%, respectively); PsARC; EULAR-defined response based on DAS28 score;23 proportion of patients in EULAR-defined remission (DAS28 score ≤2.6)31; MASES score; SPARCC score; LDI basic score; PASI score; and HRQoL by SF-36 and HAQ-DI. Post hoc analyses included treatment group comparisons of changes in CRP and ESR from baseline to week 6 and a subgroup analysis of ACR20/50/70 response for TNFα inhibitor (TNFi)-naive versus TNFi pre-exposed patients at week 6. An exploratory analysis was conducted to assess hBD-2 levels at weeks 0 and 6. Anti-drug antibody testing was performed using two validated, independent capture assays (screening and confirmation).
Statistical methods are presented in the online supplementary appendix.
Results
Patient disposition and baseline characteristics
Thirty-five (83.3%) of the 42 randomised patients completed the study (secukinumab, 25/28; placebo, 10/14) (figure 1); 3 patients (10.7%) on secukinumab and 4 patients (28.6%) on placebo discontinued prematurely (because of lack of efficacy/withdrawal of consent). Among the 28 patients randomised to secukinumab and the 14 randomised to placebo, 4 secukinumab patients (2 inclusion/exclusion criteria violations, 1 serious adverse event (SAE) and 1 disallowed medication prior to week 6) and 1 placebo patient (inclusion/exclusion criteria violations) were excluded from the efficacy analysis because of protocol violations (per-protocol analysis), leaving a total of 24 secukinumab and 13 placebo patients for the efficacy analysis. Demographics and baseline characteristics were balanced between groups, with the exception of the female to male ratio, which was close to 2:1 and thus higher than expected for a European PsA population. The majority of the patients were Caucasian (93%) (table 1). Prevalent psoriasis, prior TNFi exposure and concomitant therapy with DMARDs were reported for 23, 10 and 13 patients on secukinumab and for 11, 3 and 6 patients on placebo, respectively.
Demographics and baseline characteristics

Patient disposition. AE, adverse event.
Efficacy
The ACR20 response rate at week 6 was 39% (9/23) for patients who received secukinumab versus 23% (3/13) for those who received placebo. Although numerically higher for secukinumab, the ACR20 response rate was not statistically different from placebo (95% CI 0.38 to 15.15, p=0.27) and the primary endpoint was not met. When ACR20 and ACR50 responses were present, onset was rapid in secukinumab-treated patients, occurring as early as week 2. At week 6, higher hurdle endpoints for secukinumab versus placebo were ACR50 (17% (4/23) vs 8% (1/13), p=0.39) and ACR70 (9% (2/23) vs 0% (0/23), p=0.40) responses and PsARC (43% (10/23) vs 38% (5/13), p=0.53) (figure 2). ACR20 responses in secukinumab-treated patients were maintained over time (39% (9/23) at week 12 and 43% (10/23) at week 24/end of study).

ACR20, ACR50 and ACR70 response rates and PsARC rates over time for patients receiving secukinumab 10 mg/kg or matching placebo (two intravenous injections, 21 days apart). *Week 24 or end of study if early discontinuation; data shown in the figure are mean (SD). ACR, American College of Rheumatology; D, day; PsARC, Psoriatic Arthritis Response Criteria.
No clinically relevant differences between secukinumab- and placebo-treated patients were noted for the mean DAS28 score over time (data not shown). At week 6, good EULAR response was seen in 21.7% (5/23) of the patients treated with secukinumab versus 9.1% (1/11) treated with placebo, and at week 12, good response was seen in 33.3% (7/21) versus 18.2% (2/11) of patients, respectively. At week 6, remission (DAS28 score ≤2.6) was achieved in 21.7% (5/23) of the patients treated with secukinumab versus 9.1% (1/11) treated with placebo; at week 12, remission was achieved in 23.8% (5/21) of secukinumab-treated patients versus 9.1% (1/11) of placebo-treated patients.
A decrease in median PASI score from baseline was noted in patients treated with secukinumab up to week 24, whereas no decrease was seen in placebo patients. However, PASI scores at baseline were very low (median 2.1 for secukinumab and 2.0 for placebo), and, unsurprisingly, no meaningful interpretation of changes following secukinumab therapy was possible (see online supplementary table S1). Likewise, SPARCC and MASES enthesitis scores were low at baseline and no further decrease could be demonstrated following treatment with secukinumab (data shown in online supplementary table S1). LDI basic scores were available for 10 patients (6 on secukinumab). No or minimal changes only were observed between the mean scores at baseline and the mean scores reported for weeks 6 and 12 (see online supplementary table S1).
Results from two QoL outcome measurements are provided in table 2. Statistically significant differences between the secukinumab and the placebo groups were observed at weeks 6 and 12 for percentage change from baseline HAQ-DI score, for percentage of patients with HAQ-DI score reduction from baseline >0.3 and for the SF-36 physical component.
Summary of percentage change from baseline in HAQ-DI and SF-36 physical component scores in patients receiving secukinumab 10 mg/kg or matching placebo (two intravenous injections, 21 days apart)
Post hoc analyses
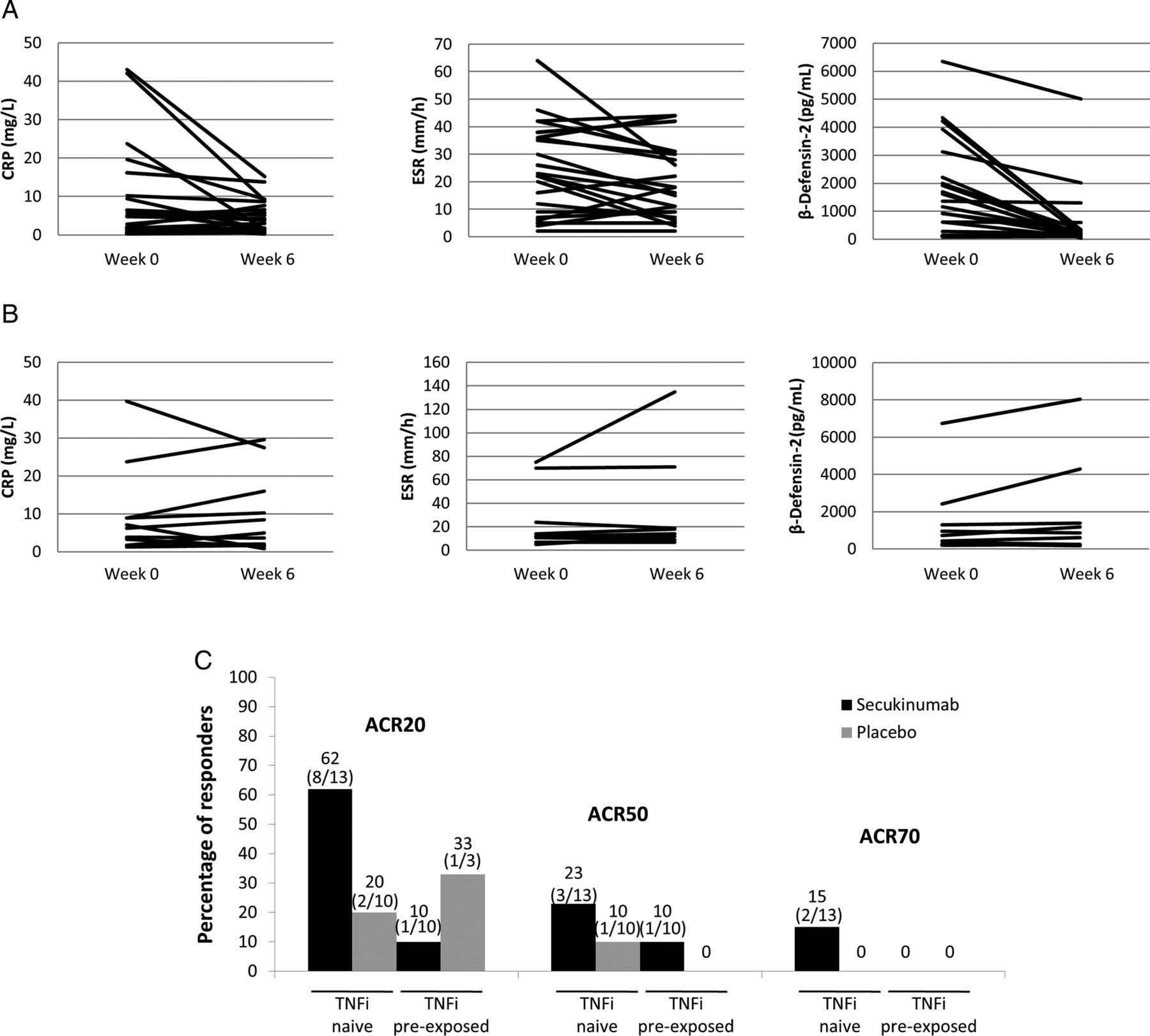
CRP and ESR changes from baseline to week 6 are depicted in figure 3A,B. Comparisons between the secukinumab and the placebo arms yielded statistically significant differences for both of these acute-phase parameters (CRP: p=0.039; ESR: p=0.038). A subgroup analysis evaluated ACR20, ACR50 and ACR70 responses at week 6 for TNFi-naive versus prior TNFi-exposed patients. Among patients with prior TNFi therapy (n=13), 1/10 (10%) patients treated with secukinumab and 1/3 (33.3%) patients treated with placebo achieved an ACR20 response. In contrast, in the TNFi-naive group (n=23), 8/13 (62%) patients treated with secukinumab achieved an ACR20 response versus 2/10 (20%) patients treated with placebo (figure 3C).
{kind=link}
{kind=link}
{kind=link}
(A) Levels of acute-phase reactants (CRP and ESR) and human β-defensin-2 (hBD-2) over time in patients receiving secukinumab 10 mg/kg (two intravenous injections, 21 days apart). (B) Levels of CRP, ESR and hBD-2 over time in patients receiving matching placebo. (C) ACR20, ACR50 and ACR70 response rates at week 6 for TNFα inhibitor (TNFi)-naive versus TNFi pre-exposed patients by treatment group. ACR, American College of Rheumatology; CRP, C reactive protein; ESR, erythrocyte sedimentation rate; TNFα, tumour necrosis factor α.
Exploratory biomarker analysis
hBD-2 concentrations were available from 34 patients (24 on secukinumab and 10 on placebo) at baseline and post-treatment, and kinetics are depicted in figure 3A,B. Marked reductions of hBD-2 serum concentrations were seen in a majority of secukinumab-treated patients, whereas only minor and random changes were noticeable for the placebo group. Reductions were most prominent for the patients with high pre-treatment hBD-2 concentrations. Comparisons between the secukinumab and the placebo arms demonstrated a highly significant advantage for secukinumab for hBD-2 reductions from baseline (p=0.0009). Summary statistics for hBD-2 at baseline and week 6 are shown in online supplementary table S2.
Safety
The rate of adverse events (AEs) was numerically higher in secukinumab versus placebo patients (26 (93%) vs 11 (79%), respectively) (table 3). Six SAEs were reported in four secukinumab patients: tendon rupture/carpal tunnel syndrome/cellulitis of the hand in one patient (the cellulitis was treated surgically and with cefuroxime 500 mg twice daily, with Staphylococcus aureus reported as the causative agent); morbid obesity in one patient (adapositas magna; diagnosed and treated with a surgical procedure during the course of the study); fall in one patient; and breast cancer in one patient (diagnosed prior to dosing, and thus constituting a protocol violation). One SAE was reported in a placebo recipient (polyarthritis; severe flare requiring hospitalisation). None of the SAEs were suspected by the investigators to be related to the study treatment.
Summary of safety events (safety population)*
There were 35 episodes of infection (26 mild, 8 moderate, 1 severe) in 23 patients (16 (57%) secukinumab patients and 7 (50%) receiving placebo). Rates and types of infections were comparable for secukinumab- and placebo-treated patients. Events of leucopenia were observed in eight secukinumab patients and one placebo patient, all Common Terminology Criteria (CTC) Grade 1; neutropenia events were observed in six secukinumab patients and one placebo patient (all CTC Grade 1 except for one: CTC Grade 2 (1.5−1.0×109/l), occurring 5 weeks after the second infusion), and no relationship with concurrent infections was apparent. No other clinically relevant alterations in laboratory parameters were reported, with the exception of a single elevated liver function test in one patient at week 24 (21 weeks after the last dosing with secukinumab). No antibodies to secukinumab were detected during the study up to week 24.
Discussion
This is the first study to examine the effects of blockade of IL-17A in a cohort of patients with active PsA. In 42 patients with moderate-to-severe PsA with or without prior TNFi exposure and with or without concomitant background DMARD therapy, secukinumab was associated with numerically greater ACR20 response rates compared with placebo, demonstrated a favourable safety profile and was well tolerated. The frequencies and types of AEs, including infections and SAEs, were comparable between the secukinumab and the placebo groups and were consistent with the safety profile seen with secukinumab in other indications, including larger studies of patients with psoriasis vulgaris and rheumatoid arthritis.15–18 ,32 While clear trends towards beneficial clinical effects of secukinumab were seen, the primary efficacy endpoint (difference in ACR20 response rates between secukinumab and placebo at week 6) was not met.
However, several observations support the view that secukinumab might exert biologically relevant effects in PsA. The rate of discontinuations was three times higher in the placebo group compared with the secukinumab group. In addition, secondary ACR response endpoints consistently showed numerically greater responses for secukinumab compared with placebo, up to and including week 24, indicating that the early effects induced by secukinumab were also durable. QoL measures demonstrated deteriorations of SF-36 physical component and HAQ-DI scores in placebo patients, as compared with secukinumab patients, who reported marked improvements over time. Although this study was not powered to detect differences in acute-phase parameters over time, a post hoc analysis showed significant differences in the reductions of CRP and ESR between secukinumab- and placebo-treated patients, particularly among secukinumab patients with elevations in pre-treatment levels. It is also worth noting that preliminary results have suggested secukinumab was effective in reducing symptoms of moderate-to-severe active ankylosing spondylitis, another form of spondyloarthritis.16
Limitations of our study that may have affected the ability to meet the primary endpoint include the small sample size and the preliminary dosing regimen of only two intravenous infusions. The study may also have been underpowered to achieve statistical significance with the chosen sample size and primary endpoint. It should also be noted that CRP levels, QoL measures, and enthesitis and PASI scores at entry were generally lower than those seen in previous studies of TNF inhibitors in combination with other treatments,33–37 suggesting that efficacy in this trial might be underestimated because of lesser disease activity or severity in our study population.
In a post hoc subgroup analysis, the ACR20 response rate at week 6 among secukinumab-treated patients in the TNFi pre-exposed subgroup was lower than that observed for placebo recipients in the TNFi pre-exposed subgroup, whereas the response rate for secukinumab among TNFi-naive patients was substantially greater than that reported for placebo recipients in either the TNFi-naive or the TNFi pre-exposed subgroup. Since the sample size of these subgroups was very small, these findings require further study in a larger patient cohort.
The notion that secukinumab treatment resulted in a biological effect in PsA patients is supported by the observed decrease in hBD-2 serum levels after active treatment with secukinumab compared with placebo; hBD-2 is an important downstream marker of the IL-17 pathway. This observation is in line with reports of hBD-2 as an important innate inflammatory marker in active plaque psoriasis13 and its top rank in the IL-17A gene signature.12 Although it was shown previously that hBD-2 mRNA is significantly downregulated in lesional skin of psoriasis patients upon blockade with etanercept14 and secukinumab,15 the relative contribution of keratinocyte-derived and, hypothetically, joint epithelium-derived hBD-2 in PsA requires further study.
A recent report suggested that the synovial tissue in PsA is enriched with IL-17R, and that the receptor IL-17RA is functionally active in PsA.8 It remains to be established whether IL-17A blockade by secukinumab has an impact on IL-17RA expression and function in the synovium. It is also possible that other IL-17 family members could play a role in mediating the PsA disease process in affected tissues; further research will be needed to determine whether secukinumab has specificity for all relevant members of this cytokine family.
Ustekinumab, an anti-IL-12/-23 p40 antibody registered for the treatment of psoriasis, showed preliminary efficacy in a phase II trial of patients with PsA.38 It is possible that the effects of ustekinumab are in part mediated by inhibition of IL-17A, which acts downstream of IL-23.39 A confirmatory phase III study of ustekinumab in active PsA is ongoing (ClinicalTrials.gov trial NCT01009086; accessed 4 December 2012).
In conclusion, secukinumab was well tolerated and demonstrated preliminary evidence for potential therapeutic benefit in patients with moderate-to-severe PsA. Larger studies are required to confirm results obtained from this initial phase II cohort.
Acknowledgments
We thank Swati Machwe, PhD, Novartis Healthcare Pvt Ltd, Hyderabad, India, for support in the writing of this manuscript. Editorial assistance was provided by BioScience Communications, New York, NY, USA, supported by Novartis Pharma AG, Basel, Switzerland.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online supplement
Footnotes
Handling editor Tore K Kvien
-
Contributors All authors meet the journal's criteria for authorship.
-
Funding This trial and publication were funded by Novartis Pharma AG, Basel, Switzerland. The sponsor was involved in the study design; in the collection, analysis and interpretation of data; in the writing of the study report; and in the decision to submit this paper for publication.
-
Competing interests IBM has served on advisory boards for Novartis. JS has served as a consultant for Novartis. JB has received honoraria for talks and advisory boards, paid consultancies and grants for studies from Abbott, Amgen, BMS, Celgene, Celltrion, Centocor, Medac, MSD (Schering-Plough), Novartis, Pfizer (Wyeth), Roche, Sanofi-Aventis and UCB. PE has served as an investigator in clinical trials and provided expert advice for Abbott, BMS, MSD, Novartis, Pfizer, Roche and UCB. DvdH has received consulting fees and/or research grants from Abbott, Amgen, AstraZeneca, BMS, Centocor, Chugai, Eli Lilly, GSK, Merck, Novartis, Otsuka, Pfizer, Roche, Sanofi-Aventis, Schering-Plough, UCB and Wyeth. JDI has no conflicts to declare. GD has no conflicts to declare. JW has served on advisory boards and speakers bureaus for Novartis. HS-K has no conflicts to declare. JK has served as a statistical consultant to Novartis. SM is an employee of and owns stock in Novartis. MMS is an employee of Novartis. APB is an employee of and owns stock in Novartis. WH is an employee of and owns stock in Novartis. P-PT has served on advisory boards for Novartis.
-
Patient consent Obtained.
-
Ethics approval Independent Ethics Committee or Institutional Review Board.
-
Provenance and peer review Not commissioned; externally peer reviewed.