Article Text

Abstract
Objectives Epigenetic modifications such as DNA methylation and histone acetylation have been implicated in the pathogenesis of systemic sclerosis. However, histone methylation has not been investigated so far. We therefore aimed to evaluate the role of the trimethylation of histone H3 on lysine 27 (H3K27me3) on fibroblast activation and fibrosis.
Methods H3K27me3 was inhibited by 3-deazaneplanocin A (DZNep) in cultured fibroblasts and in two murine models of dermal fibrosis. Fibrosis was analysed by assessment of the dermal thickening, determination of the hydroxyproline content and by quantification of the numbers of myofibroblasts. The expression of fos-related antigen 2 (fra-2) was assessed by real-time PCR, western blot and immunohistochemistry and modulated by siRNA.
Results Inhibition of H3K27me3 stimulated the release of collagen in cultured fibroblasts in a time and dose-dependent manner. Treatment with DZNep exacerbated fibrosis induced by bleomycin or by overexpression of a constitutively active transforming growth factor β receptor type I. Moreover, treatment with DZNep alone was sufficient to induce fibrosis. Inhibition of H3K27me3 induced the expression of the profibrotic transcription factor fra-2 in vitro and in vivo. Knockdown of fra-2 completely prevented the profibrotic effects of DZNep.
Conclusions These data demonstrate a novel role of H3 Lys27 histone methylation in fibrosis. In contrast to other epigenetic modifications such as DNA methylation and histone acetylation, H3 Lys27 histone methylation acts as a negative regulator of fibroblast activation in vitro and in vivo by repressing the expression of fra-2.
- Fibroblasts
- Systemic Sclerosis
- Inflammation
Statistics from Altmetric.com
Introduction
Systemic sclerosis (SSc) is a connective tissue disease that primarily affects the skin, but also the lungs, heart and gastrointestinal tract.1 The most obvious hallmark of SSc is the massive accumulation of extracellular matrix components in involved organs. The resulting tissue fibrosis often results in failure of the affected organs and is a major cause of the high morbidity and increased lethality in SSc patients. Fibrosis is caused by an excessive release of extracellular matrix by aberrantly activated fibroblasts.2 However, the molecular mechanisms of the pathological activation of SSc fibroblasts are incompletely understood.
Epigenetic modifications are defined as alterations of the chromatin structure without changes in the nucleotide sequence and are important regulators of transcription.3 ,4 Deregulated epigenetic alterations have been linked to various human diseases and also contribute to the pathogenesis of SSc.5 ,6 Aberrant DNA methylation as well as uncontrolled activation of histone deacetylases (HDAC) have been demonstrated in SSc and contribute to the activated phenotype of SSc fibroblasts.7–9 In contrast to DNA methylation and histone acetylation, the role of histone methylation in fibrotic diseases has not yet been investigated. In particular, trimethylation of histone H3 on lysine 27 (H3K27me3) is a common epigenetic modification that potently represses the transcription of target genes. H3K27me3 is mediated by so-called polycomb group proteins such as enhancer of zeste 2 (EZH2), suppressor of zeste 12 (SUZ12) and embryonic ectoderm development that act together in polycomb repressive complexes.10 ,11 The methylation of histones influences transcription, by providing a binding surface for several chromatin remodelling enzymes with specific methyl-lysine binding sites, by blocking the binding of proteins that interact exclusively with unmethylated histones or by inhibition of the catalyses of other neighbouring residues. As for other epigenetic modifications, histone methylation is reversible and can be targeted therapeutically. 3-Deazaneplanocin A (DZNep) has been identified as a potent inhibitor of polycomb repressive complexes that abrogates H3K27me3.12 ,13 DZNep has shown promising effects in preclinical cancer models and is currently being evaluated in clinical trials for the treatment of various types of cancer.14 Given the role of epigenetic modifications in the pathogenesis of SSc, the potent regulatory effects of H3K27me3 on gene transcription and the availability of DZNep for therapy, we aimed to evaluate the role of H3K27me3 for fibroblast activation and fibrosis in the present study.
Materials and methods
Patients and fibroblast cultures
Human fibroblasts from SSc patients and healthy individuals were prepared by outgrowth cultures from skin biopsy specimens and cultured as described.15 SSc fibroblast cultures were obtained from skin biopsies of clinically involved skin (n=8). All patients had diffuse cutaneous SSc. The median age of SSc patients was 36 years (range 19–61 years) and their median disease duration was 5 years (range 0.5–9 years). All patients were positive for antinuclear antibodies; four patients were positive for anti-topoisomerase-1 antibodies, but no patient was positive for anti-centromere antibodies. Skin fibrosis was progressive in four patients at the time of the biopsy, whereas the other four patients had stable skin fibrosis. None of the patients was treated with immunosuppressive or other potentially disease-modifying drugs at the time of biopsy. All patients and controls signed a consent form approved by the local institutional review boards. Fibroblasts from passages 4 to 9 were used for the experiments.
Inhibition of H3K27me3 in dermal fibroblasts
Stimulation experiments were performed in DMEM/0.1% fetal calf serum for 4 days. Dermal fibroblasts were incubated with DZNep in concentrations ranging from 0.2 to 5.0 µmol/l for 48 h. For a concentration of 5 µmol/l, the incubation time varied from 6 h to 48 h. DZNep was dissolved in phosphate-buffered saline. These concentrations cover the mean peak and trough concentrations achieved in human cancer trials.
Quantitative real-time PCR
Gene expression was quantified by SYBR green real-time PCR using the ABI Prism 7300 Sequence Detection System (Applied Biosystems, Foster City, California, USA).16 ,17 Specific primer pairs for each gene were designed with the Primer 3 software. The following primer pairs were used for the analyses: for α1(I) procollagen (col 1a1), 5′–ACGAAGACATCCCACCAATC–3′ (forward) and 5′–ATGGTACCTGAGGCCGTTC–3′ (reverse); for α-smooth muscle actin, 5′–AAGAGGAATCCTGACCCTGAA–3′ (forward) and 5′–TGGTGATGATGCCATGTTCT–3′ (reverse); for fos-related antigen 2 (Fra-2), 5′–AGCTGGAGGAGGAGAAGTCA–3′ (forward) and 5′–CTGCAGCTCAGCAATCTCC–3′ (reverse). Samples without enzyme in the room temperature (RT) reaction (non-RT controls) were used as negative controls. Unspecific signals caused by primer dimers were excluded by no-template controls and by dissociation curve analysis. β-Actin (Applied Biosystems) was used as a housekeeping control to normalise for the amounts of complementary DNA within each sample. Differences were calculated with the threshold cycle and the comparative threshold cycle method for relative quantification.
Western blot analysis
Protein extracts were prepared as described;18 10 µg of protein were separated by sodium dodecylsulphate–polyacrylamide gel electrophoresis and electrotransferred onto polyvinylidene difluoride membranes (Roth, Karlsruhe, Germany) according to standard protocols.19 Fra-2 was detected using polyclonal rabbit-anti-human Fra-2 antibodies (Santa Cruz Biotechnology, Heidelberg, Germany) and horseradish peroxidase (HRP)-conjugated polyclonal goat anti-rabbit antibodies (DAKO, Hamburg, Germany). H3K27 methylation was detected using monoclonal rabbit-anti H3K27 antibodies (Cell Signalling, Life Technologies, Darmstadt, Germany) and HRP-conjugated polyclonal goat anti-rabbit antibodies (DAKO). Equal loading of proteins was confirmed by visualisation of Lamin A/C (New England BioLabs, Frankfurt, Germany). The intensity of the bands was quantified using ImageJ Software (V.1.41).
Collagen measurements
The collagen content in cell culture supernatants was analysed with the hydroxyproline assay and SirCol collagen assay (Biocolor, Belfast, Northern Ireland) as described.20
Nucleofection with siRNA against Fra-2
Dermal fibroblasts were transfected with 2.5 µg of specific small interfering RNA duplexes against Fra-2 using the human dermal fibroblast Nucleofector Kit (Lonza, Basel, Switzerland).21 Fibroblasts transfected with non-targeting control siRNA (Ambion, Darmstadt, Germany) served as controls.
Immunohistochemistry for Fra-2
The expression of Fra-2 was detected by staining with polyclonal mouse-anti-human Fra-2 antibodies (ab15296; Abcam, Cambridge, UK), polyclonal goat anti-mouse antibodies (Jackson ImmunoResearch, Soham, UK) labelled with HRP and 3,3′diaminobenzidine tetradydrochloride (DAB, Merck, Darmstadt, Germany).
Treatment of mice with DZNep
Bleomycin-induced experimental fibrosis
Skin fibrosis was induced in 6-week-old, pathogen-free, male DBA 2J mice (Janvier, Le Genest-Saint-Isle, France) by the injection of bleomycin as previously described.22 Subcutaneous injections of 100 μl 0.9% NaCl, the solvent for bleomycin, were used as controls. To investigate the effect of the inhibition of H3K27me3 on experimental fibrosis, one group of eight mice was treated for 3 weeks with DZNep at doses of 2 mg/kg twice per week by intraperitoneal injections. After 21 days, animals were killed by cervical dislocation. Eight mice per group were analysed.
Dermal fibrosis induced by overexpression of a constitutively active TGF-β receptor I construct
Dermal fibrosis was induced in pathogen-free C57BL/6 mice (Janvier) by intracutaneous injections of attenuated type V adenoviruses overexpressing a constitutively active transforming growth factor β (TGFβ) receptor I (AdTBR) as described.23 Four mice infected with AdTBR were treated with DZNep at doses of 2 mg/kg twice a week by intraperitoneal injections. Another four mice infected with AdTBR were sham treated with 0.9% NaCl, the solvent of DZNep. Four mice infected with attenuated type V adenoviruses encoding only for LacZ and sham treated with 0.9% NaCl served as controls.
Treatment of mice with DZNep in the absence of profibrotic stimuli
To evaluate whether the inhibition of H3K27me3 alone is sufficient to induce skin fibrosis, C57BL/6 mice were treated with DZNep at doses of 2 mg/kg twice a week by intraperitoneal injections for up to 8 weeks.
Quantification of experimental dermal fibrosis
For both mouse models, dermal fibrosis was quantified by measuring dermal thickness, hydroxyproline content and the number of myofibroblasts as described previously.21 ,24 ,25 The local ethics committee approved all animal experiments.
Statistical analysis
Data are expressed as the median±IQR. Differences between the groups were tested for their statistical significance by the Mann–Whitney U non-parametric test. p Values are expressed as follows: 0.05>p>0.01 as *; 0.01>p>0.001 as **; p<0.001 as ***.
Results
Inhibition of histone methylation by DZNep stimulates the release of collagen in cultured fibroblasts
First, we analysed, whether H3K27me3 is altered in SSc fibroblasts. We found that H3K27me3 is increased in SSc fibroblasts as compared to fibroblasts from healthy volunteers (figure 1A). We next study the role of H3K27me3 in fibroblast activation using the H3K27me3 inhibitor DZNep. Incubation with DZNep upregulated the messenger RNA levels of col 1a1 in SSc fibroblasts a time dependent manner with a peak after 12 h (figure 1B). The stimulatory effects of DZNep on col 1a1 mRNA were also dose dependent in concentrations ranging from 0.2 to 5.0 μM with maximal increases at concentrations of 5.0 μM (figure 1C). Consistently, incubation with DZNep dose-dependently increased the amount of collagen protein in supernatant of dermal fibroblasts (figure 1D). DZNep did not significantly increase the mRNA levels of other extracellular matrix proteins such as fibronectin.
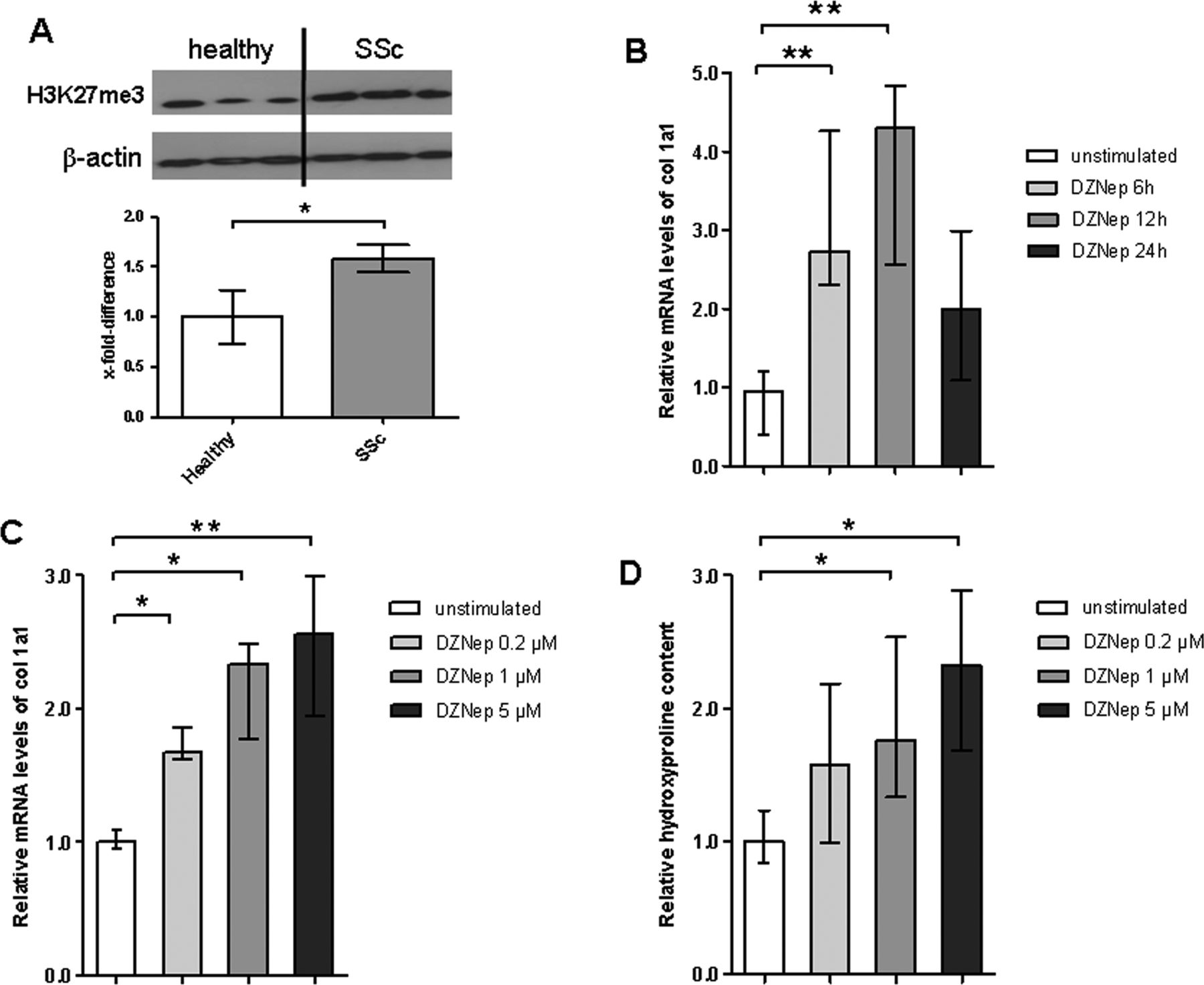
The trimethylation of histone H3 on lysine 27 (H3K27me3) regulates the release of collagen from fibroblasts. (A) H3K27me3 is increased in cultured systemic sclerosis (SSc) fibroblasts compared to fibroblasts from healthy individuals. Representative examples from a total of five SSc fibroblast cultures and seven fibroblast lines from healthy individuals are shown. (B) Incubation of SSc fibroblasts with 3-deazaneplanocin A (DZNep) increases the mRNA levels of α1(I) procollagen (col 1a1) in a time-dependent manner with maximal effects after 12 h (n=8). (C) DZNep dose-dependently upregulates the mRNA levels of col 1a1 with maximal effects observed at a concentration of 5.0 μM (n=8). (D) DZNep upregulates the release of collagen protein in a dose-dependent manner as shown by hydroxyproline assays (n=8).
Treatment with DZNep exacerbates bleomycin-induced dermal fibrosis
After demonstrating that inhibition of H3K27me3 stimulates collagen synthesis in vitro, we next analysed the effects of DZNep on bleomycin-induced dermal fibrosis, a model resembling early, inflammatory stages of SSc.26 Mice injected with bleomycin developed dermal fibrosis with a massive accumulation of thickened collagen bundles. However, inhibition of histone methylation further exacerbated bleomycin-induced dermal fibrosis (figure 2A). The dermal thickening in bleomycin-challenged mice treated with DZNep was significantly more pronounced compared to sham-treated mice challenged with bleomycin (figure 2B). Treatment with DZNep also significantly enhanced myofibroblast counts (figure 2C) and the hydroxyproline content (figure 2D) compared to bleomycin alone.

Inhibition of the trimethylation of histone H3 on lysine 27 exacerbates bleomycin induced dermal fibrosis. (A) Representative sections of mice injected with NaCl, bleomycin and bleomycin plus 3-deazaneplanocin A (DZNep) are shown at 100-fold magnifications. (B) Increased dermal thickening in bleomycin-challenged mice treated with DZNep compared to bleomycin-challenged mice receiving sham treatment. (C) and (D) Increased myofibroblast counts and enhanced hydroxyproline content in the skin of mice injected with DZNep and bleomycin. n=8 for all groups.
Inhibition of histone methylation enhances fibrosis in the AdTBR model
We next investigated the effects of DZNep in the AdTBR model, which serves as a model of later, less inflammatory stages of SSc.26 Injection of AdTBR into the skin of mice induced localised fibrosis with dermal thickening, accumulation of collagen and differentiation of resting fibroblasts into myofibroblasts compared to control mice injected with AdLacZ (figure 3A–D). Inhibition of histone methylation by DZNep further exacerbated AdTBR-induced fibrosis (figure 3A). Dermal thickening was enhanced compared to sham-treated AdTBR mice (p<0.05) (figure 3B). The number of myofibroblasts and the hydroxyproline content were also significantly higher in AdTBR mice treated with DZNep compared to sham-treated AdTBR mice (figure 3C,D).
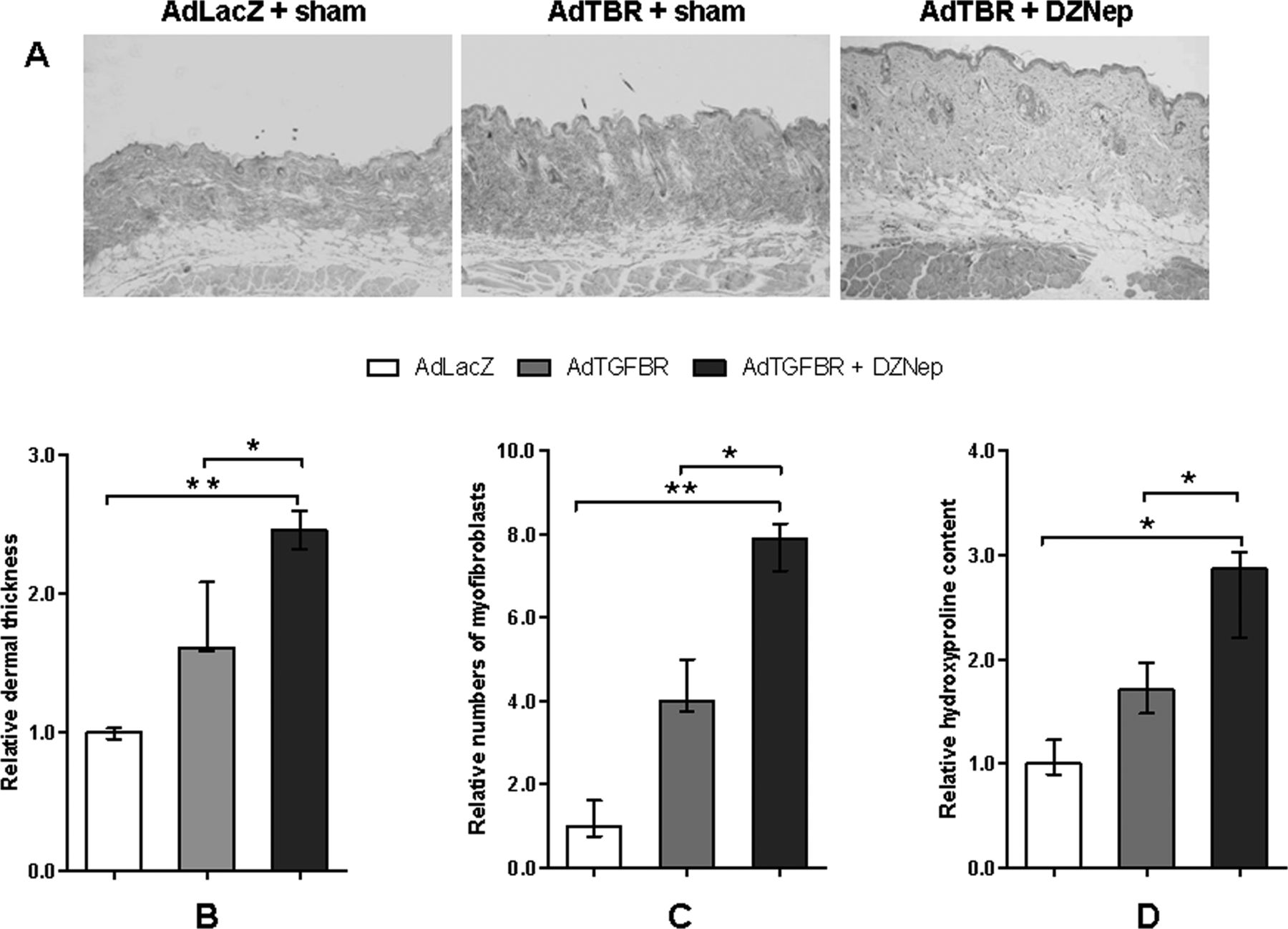
The trimethylation of histone H3 on lysine 27 (H3K27me3) inhibitor 3-deazaneplanocin A (DZNep) increases fibrosis in the attenuated type V adenoviruses overexpressing a constitutively active transforming growth factor β receptor I (AdTBR) model. (A) Representative sections of mice injected with AdLacZ, AdTBR and AdTBR plus DZNep are shown at 100-fold magnifications. (B)–(D) Inhibition of H3K27me3 promotes dermal thickening, stimulates the differentiation of resting fibroblasts into myofibroblasts and increases the hydroxyproline content. n=4 for all groups.
Treatment with DZNep alone induces skin fibrosis
To investigate whether the inhibition of H3K27me3 alone is sufficient to induce skin fibrosis, wild-type mice were treated with DZNep in the absence of additional profibrotic stimuli. The first evidence of fibrosis was detectable within 4 weeks of treatment and progressed further with significant increases in dermal thickness, myofibroblast counts and hydroxyproline content after 8 weeks of treatment with DZNep (figure 4A–D).

Inhibition of the trimethylation of histone H3 on lysine 27 (H3K27me3) alone is sufficient to induce fibrosis. (A) Representative sections of mice treated with 3-deazaneplanocin A (DZNep) for either 4 weeks or 8 weeks and sham-treated control mice are shown at 100-fold magnifications. (B), (C) and (D) Inhibition of H3K27me3 induces dermal thickening, promotes myofibroblast differentiation and increases the hydroxyproline content.
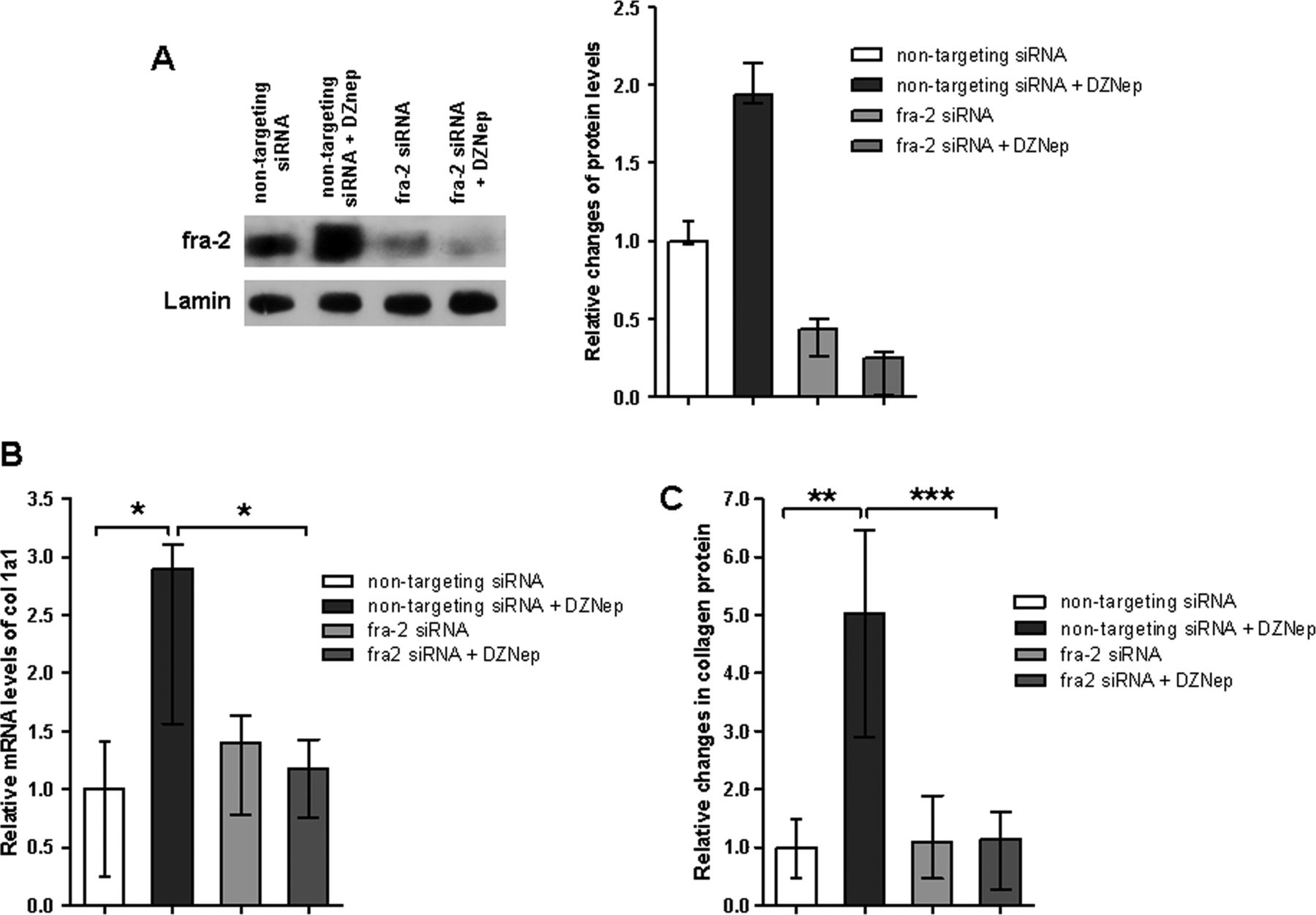
Inhibition of H3K27me3 exerts its profibrotic effects by induction of fra-2
To identify the molecular mechanism underlying the induction of collagen by DZNep, we screened fibroblasts incubated with DZNep for differentially expressed profibrotic genes. We found that DZNep induces the expression of fra-2, a transcription factor of the AP-1 family that has recently been implicated in the pathogenesis of SSc.27 ,28 Treatment of cultured fibroblasts with DZNep potently induced the mRNA levels of fra-2 with maximal increases after 24 h (figure 5A). Similar results were obtained on the protein level (figure 5B). We next confirmed the induction of fra-2 in vivo in mice treated with DZNep. Consistent with previous reports, challenge with bleomycin or injection of AdTBR increased the expression of fra-2 compared to non-fibrotic control mice. However, treatment with DZNep further increased the numbers of fibroblasts expressing fra-2 protein in both models (figure 5C,D). DZNep also increased the mRNA of connective tissue growth factor (CTGF) in cultured fibroblasts, although the induction was not as pronounced as for fra-2. We also did not detect significant differences in the levels of CTGF in DZNep-treated mice (data not shown).

3-Deazaneplanocin A (DZNep) induces the expression of the profibrotic transcription factor fos-related antigen 2 (fra-2). (A) and (B) Treatment of human fibroblasts with DZNep increases the mRNA and protein levels of the AP-1 transcription factor fra-2 (n=5 for mRNA and n=3 for protein). (C) Treatment with DZNep upregulates the number of fra-2-positive fibroblasts in bleomycin-induced dermal fibrosis (n=8) and (D) in mice injected with attenuated type V adenoviruses overexpressing a constitutively active transforming growth factor β receptor I (AdTGFBR) (n=4).
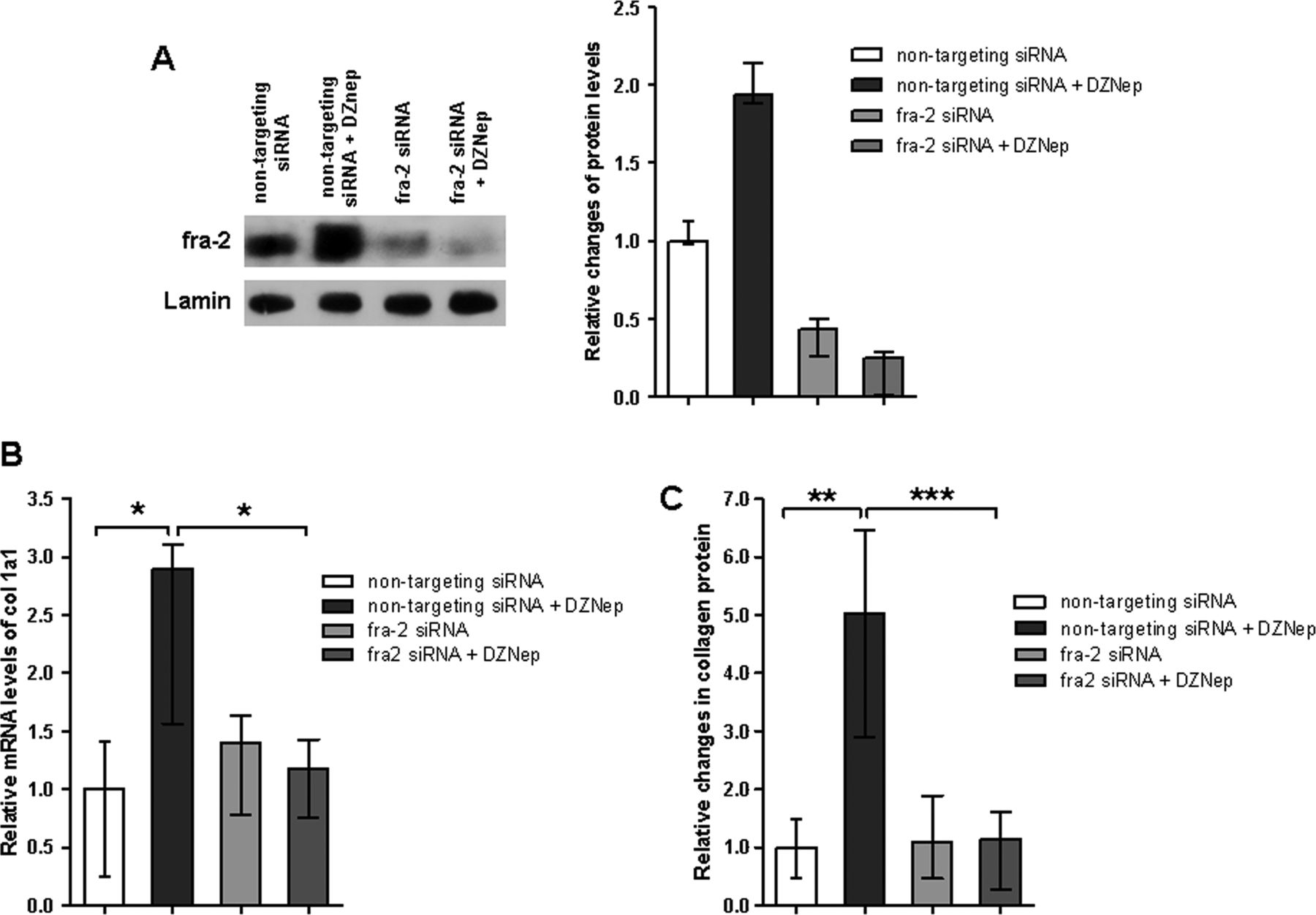
To investigate the functional role of fra-2, we analysed whether knockdown of fra-2 ameliorates the profibrotic effects of DZNep. siRNA against fra-2 effectively decreased the expression of fra-2 in fibroblasts (figure 6A). Knockdown of fra-2 decreased the basal release of collagen from cultured fibroblasts. Moreover, knockdown of fra-2 completely prevented the stimulatory effects of DZNep on col 1a1 mRNA levels (figure 6B) and on collagen protein (figure 6C), demonstrating that profibrotic effects of DZNep are mediated by fra-2.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The profibrotic effects of 3-deazaneplanocin A (DZNep) are mediated by fos-related antigen 2 (fra-2). (A) Western blot analyses demonstrated a strong decrease in the levels of fra-2 protein in cultured fibroblasts by fra-2 siRNA (n=3). (B) and (C) Knockdown of fra-2 in cultured fibroblasts by siRNA completely abrogated the stimulatory effects of DZNep on α1(I) procollagen mRNA (n=6) and collagen protein (n=6).
Discussion
Different epigenetic modifications have recently emerged as important regulators of fibroblast activation, and therapeutic approaches targeting aberrant epigenetic modifications have been suggested as potential therapies for fibrotic diseases.5 The role of DNA methylation is characterised in most detail. Antifibrotic genes such as fli-1 are silenced by promoter hypermethylation in SSc fibroblasts and re-activation of these genes abrogates the activated phenotype of SSc fibroblasts.9 Moreover, treatment of mice with 5-aza, an inhibitor of DNA methyltransferases reduces fibrosis in different experimental models.29 In addition to targeting DNA methylation, HDAC inhibitors may also offer therapeutic potential. HDAC inhibitors decreased the release of collagen from cultured fibroblasts and ameliorate bleomycin-induced fibrosis.7 ,8 As for inhibitors of DNA methyltransferases, the proposed primary mechanism is the induction of antifibrotic genes rather than inhibition of profibrotic genes. We demonstrate in the present study a potential role of H3K27me3 in the pathogenesis of SSc. Moreover, He et al30 demonstrated recently that DZNep arrests experimental graft versus host disease by activating the expression of the pro-apoptotic gene Bim selectively in autoreactive T cells. Similar to the effects of DNA methyltransferase inhibitors and HDAC inhibitors, treatment with DZNep removes inhibitory epigenetic signals and activates the transcription of target genes. However, the inhibition of H3K27me3 has opposite effects on fibroblast activation. Inhibitors of DNA methyltransferases and HDAC inhibitors have antifibrotic effects, whereas inhibition of H3K27me3 stimulates the release of collagen from cultured fibroblasts, induces fibrosis in the absence of profibrotic stimuli and exacerbates bleomycin and TBR-induced experimental fibrosis. In contrast to the profibrotic effects of DNA methyltransferases and histone deactylases, H3K27me3 thus acts as a negative regulator to prevent aberrant fibroblast activation and fibrosis. The upregulation of H3K27me3 in SSc fibroblasts may thus be a compensatory attempt to counterregulate the increased expression of profibrotic genes in SSc fibroblasts.
We identified the AP-1 transcription factor fra-2 as a potential molecular mediator of the profibrotic effects of the inhibition of H3K27me3. Treatment with DZNep increases the mRNA and protein levels of fra-2 in cultured fibroblasts as well as in two different murine models of fibrosis. Moreover, knockdown of fra-2 prevents the profibrotic effects of DZNep in vitro, indicating that the stimulatory effects on fibroblasts on the inhibition of H3K27me3 may depend on fra-2. Of note, fra-2 has recently been identified as a key player in the pathogenesis of SSc.27 ,28 Fra-2 is overexpressed in SSc and regulates the release of collagen from fibroblasts. Mice overexpressing fra-2 develop a rapidly progressive systemic fibrotic disease that invariable leads to death within 5 months, thereby highlighting the potent profibrotic effects of fra-2.31 However, inhibition of H3K27me3 will not selectively affect the expression of fra-2, but will also modulate the expression of other target genes, and the altered expression of those genes probably contributes to the profibrotic effects of DZnep. A potential additional candidate is IP-10 (inducible protein 10 kDa), which has recently been found to be hypermethylated in cultured fibroblasts from patients with idiopathic pulmonary fibrosis.32
In summary, we demonstrate that the inhibition of H3K27me3 by DZNep stimulates the release of collagen from fibroblasts and exacerbates bleomycin as well as TBR-induced experimental fibrosis. We identified fra-2 as one potential mediator of these profibrotic effects. Our data demonstrate that H3K27me3, in contrast to other epigenetic changes, acts as a negative regulator of tissue fibrosis. However, further studies are required to elucidate the precise role of H3K27me3. These experiments may include additional studies on the molecular mechanism of transcriptional regulation by H3K27me3, further identification of target genes, factors that regulate histone methylation in fibroblasts and also additional studies on other animal models that mimic different aspects of SSc. In particular, the effects of H3K27me3 on vascular disease in SSc remain to be determined. It will also be interesting to determine the role of histone methylation in other fibrotic disorders of the skin such as in systemic nephrogenic fibrosis or hypertrophic scars and organ fibrosis. Moreover, other histone methylation marks in addition to H3K27me3 might also control fibroblast activation. H3K4 methylation and H3K9 methylation seem potentially interesting as they have recently been shown to be changed in response to TGFβ in rat mesangial cells.33
Acknowledgments
The authors would like to thank Maria Halter, Anna-Maria Herrmann, Madeleine Demleitner, Verena Wäsch and Stefan Fritz for excellent technical assistance.
References
Footnotes
Contributors Design of the study: MK, CD, VEM, OD, GS, JHWD. Acquisition of data: MK, CD, JH, IS, KPZ, PZ, KG, CB, AD. Interpretation of data: MK, CD, CB, AD, OD, GS, JHWD. Manuscript preparation: OD, JHWD. Provision of essential tools: KG, VEM.
Funding This study was funded by grants DI 1537/1–1, DI 1537/2-1, DI 1537/4-1, DI 1537/5-1, AK 144/1-1 and SCHE 1583/7-1 of the Deutsche Forschungsgesellschaft, grants A20 and A40 of the IZKF in Erlangen, the ELAN-Programme of the University of Erlangen-Nuremberg and the Career Support Award of Medicine of the Ernst Jung Foundation. This research was supported in part by the Intramural Research Programme of the NIH, National Cancer Institute, Center for Cancer Research.
Competing interests None.
Ethics approval Ethics approval was received from the Ethics Committee of the University of Erlangen-Nuremberg.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.