Article Text

Abstract
Objectives Millions of patients worldwide are treated with therapeutic monoclonal antibodies. These biological therapeutics can be immunogenic, resulting in anti-drug antibody formation which leads to loss of response. Fully human biological agents, such as the anti-tumour necrosis factor α (anti-TNFα) antibody adalimumab, are considered to be weakly immunogenic, but anti-adalimumab antibodies (AAA) were recently detected in more than half of treated patients with rheumatoid arthritis (RA) within 28 weeks of treatment. A study was undertaken to determine the mechanism by which AAA lead to loss of response.
Methods The specificity of the repertoire of AAA was investigated in a cohort of 50 AAA-positive RA patients. Inhibition experiments using TNFα and patient-derived anti-adalimumab monoclonal antibodies were performed.
Results The antibody response against adalimumab is highly restricted: Fab fragments of a single monoclonal antibody specific for the idiotype of adalimumab inhibited 98.65% (25th–75th percentiles: 98.25–99.90) of the total anti-adalimumab reactivity in serum from 50 AAA-positive patients. The anti-adalimumab response was confined to the TNFα binding region of adalimumab, thereby neutralising its therapeutic efficacy. In line with this restricted specificity, small immune complexes were found in the circulation of AAA-forming patients.
Conclusions The humoral immune response against adalimumab is highly restricted and limited to the idiotype of the therapeutic antibody. All antibodies result in functional neutralisation of the drug, thereby providing a mechanism by which AAA formation leads to clinical non-response.
Statistics from Altmetric.com
Introduction
The use of therapeutic monoclonal antibodies has revolutionised the treatment of many diseases. In recent years, millions of patients have been successfully treated with these biological agents. However, long-term treatment with therapeutic monoclonal antibodies can induce anti-drug antibody (ADA) formation that is associated with lower drug levels and clinical non-response.1–4 The mechanism by which the formation of anti-adalimumab antibodies (AAA) hampers clinical response is still unknown. We hypothesise two possible mechanisms. First, administration of a drug to patients producing AAA leads to the formation of immune complexes which might result in accelerating clearance of the drug. Second, neutralising AAAs might block the binding of the drug to its target.5
The tumour necrosis factor α (TNFα) blocking therapeutic antibody adalimumab is widely used in different inflammatory diseases such as rheumatoid arthritis (RA), ankylosing spondylitis and Crohn's disease.6–8 Some patients who are chronically treated with adalimumab generate antibody responses against this therapeutic monoclonal antibody which are linked to low functional drug levels and reduced clinical response.3 ,9–14 The percentage of patients developing AAA is underestimated in these studies since, in all standard assays, the measurement of AAA is hampered by the presence of the drug itself. We recently developed a novel assay enabling measurement of AAA in the presence of drug.15 Using this assay, we have shown that more than half of adalimumab-treated patients with RA produce AAA in the first 28 weeks of treatment. In most of these patients, however, this does not lead to clinical non-response since only 22% of patients fail treatment at this time point. To obtain a better insight into the mechanisms by which AAA formation influences treatment efficacy and safety, more knowledge is required on the specificity and the immunological consequences of AAA against adalimumab.
According to the anti-idiotype theory presented by Niels Jerne in the 1970s, the variable region of a given antibody contains several distinct sites (idiotopes) that together form the idiotype, against which a variety of anti-idiotypic antibody molecules can be formed.16 ,17 Idiotopes may be located at the actual antigen binding site of the recognised antibody and may also include variable region sequences outside of the antigen binding site. Depending on the idiotopes recognised, anti-idiotype antibodies may or may not block the interaction of the antibody with its target antigen.
In this study we have investigated the mechanism by which the AAA response against adalimumab leads to clinical non-response by studying the diversity of epitopes involved in the antibody response against adalimumab and the extent to which these are located on the antigen binding part of adalimumab, thus blocking binding to TNFα.
Materials and methods
See online supplementary data.
Results
Isolation and characterisation of anti-adalimumab-producing B cells
In order to investigate the mechanism by which AAA lead to clinical non-response, we first studied the number of epitopes involved in the immune response against adalimumab. We therefore obtained adalimumab-specific monoclonal antibodies. Using these antibodies as a tool in subsequent inhibition studies, we aimed at mapping the anti-adalimumab response. To obtain AAA, adalimumab-specific B cells isolated from an adalimumab-treated patient were sorted by flow cytometry. Six positive clones (as tested by measuring supernatant in the bridging ELISA) were obtained from the CD27 B cell population from which RNA was extracted (see supplementary online table S1). Two antibodies were recombinantly expressed leading to two immunoglobulin G1 (IgG1) monoclonal antibodies designated anti-adalimumab 1.1 (originally identified as an IgG1 antibody) and anti-adalimumab 1.2 (originally identified as an IgG4 antibody). Both monoclonal antibodies underwent extensive somatic hypermutation and originated from different precursor B cells (see supplementary online table S2). Both monoclonal antibodies specifically bound to adalimumab and neither showed cross-reactivity with infliximab (another therapeutic anti-TNFα monoclonal antibody) (figure 1A).

Anti-adalimumab 1.1 and anti-adalimumab 1.2 compete for binding to adalimumab. (A) Detection of increasing amounts of anti-adalimumab 1.1 and anti-adalimumab 1.2 in a bridging ELISA for the measurement of anti-adalimumab and a bridging ELISA for the measurement of anti-infliximab. (B) Increasing amounts of anti-adalimumab 1.2 Fab inhibit binding of adalimumab-specific anti-adalimumab antibodies (AAA) in patient serum but not infliximab-specific ADA tested in the antigen binding test. (C) Titration of biotinylated anti-adalimumab 1.1 and anti-adalimumab 1.2 in the absence or presence of 12.5 µg/ml unlabelled anti-adalimumab 1.1 or 12.5 µg/ml unlabelled anti-adalimumab 1.2. (D) Inhibition of biotinylated anti-adalimumab by non-biotinylated anti-adalimumab is dose-dependent.
The antibody response to adalimumab in patients with RA is restricted to the idiotype
We investigated how representative these two antibodies are for the total antibody repertoire found in patient serum. First, an inhibition study was carried out with serum from the patient who served as B cell donor for the generation of recombinant antibodies. Remarkably, in an antigen binding test (ABT) specific for anti-idiotype antibodies, Fab fragments of anti-adalimumab 1.2 prevented 99% of the adalimumab-specific reactivity from binding to radiolabelled adalimumab F(ab′)2 (figure 1B), showing that the total antibody response in this patient is directed against the same region as anti-adalimumab 1.2. In line with this finding, binding of either biotinylated anti-adalimumab 1.1 or 1.2 to adalimumab could be dose-dependently inhibited by unlabelled anti-adalimumab 1.1 as well as anti-adalimumab 1.2 (figure 1C,D), indicating that both antibodies bind to overlapping epitopes on adalimumab. In contrast, binding of anti-infliximab antibodies to infliximab F(ab′)2 was not influenced by addition of anti-adalimumab 1.2 Fab fragments (figure 1B). Next, nine more anti-adalimumab-positive patients were tested in the same assay (figure 2A). In all cases the antibody response to adalimumab could be at least 98% inhibited by anti-adalimumab 1.2 Fab. This indicates that a single monoclonal Fab fragment is able to inhibit all anti-idiotype antibodies present in patient serum, suggesting that the entire AAA response in these patients is directed against the same or overlapping epitopes of adalimumab.

Binding of serum anti-adalimumab antibodies (AAA) of 50 patients to adalimumab is inhibited by anti-adalimumab 1.2. (A) Ten patient serum samples containing AAA against adalimumab were inhibited by the addition of increasing amounts of anti-adalimumab 1.2 Fab in the antigen binding test (ABT). The results were normalised to the binding percentage of the non-inhibited sample. Binding was significantly (p<0.0001) inhibited by the addition of 2 µg Fab fragments (paired t test) (B) Five representative patient serum samples containing AAA are inhibited by Fab fragments of anti-adalimumab 1.2 as tested in the bridging ELISA. Again binding was significantly (p<0.0001) inhibited by the addition of 2 µg Fab fragments (paired t test). (C) The percentage inhibition in the total of 50 patients tested in either the ELISA or ABT (mean±SD 98.52±0.99).
In a larger group of 40 patients a similar inhibition assay was carried out using a bridging ELISA that also allows detection of antibodies against the constant region of adalimumab. Pre-incubation of the conjugate with Fab fragments of anti-adalimumab 1.2 again resulted in 94–99% inhibition of the signal (figure 2B), while pre-incubation with irrelevant human IgG Fab fragments had no effect (data not shown). Thus, the antibody response in the serum of a total of 50 anti-adalimumab-positive patients tested either by the ABT (10 patients) or the bridging ELISA (40 patients) could be 94–99% inhibited (median 98.65%; 25th–75th percentiles: 98.25–99.90%; figure 2C) by a single monoclonal antibody (anti-adalimumab 1.2). This demonstrates that the antibody response against adalimumab is highly restricted. The vast majority of the antibodies bind to overlapping epitopes.
All antibodies against adalimumab are neutralising
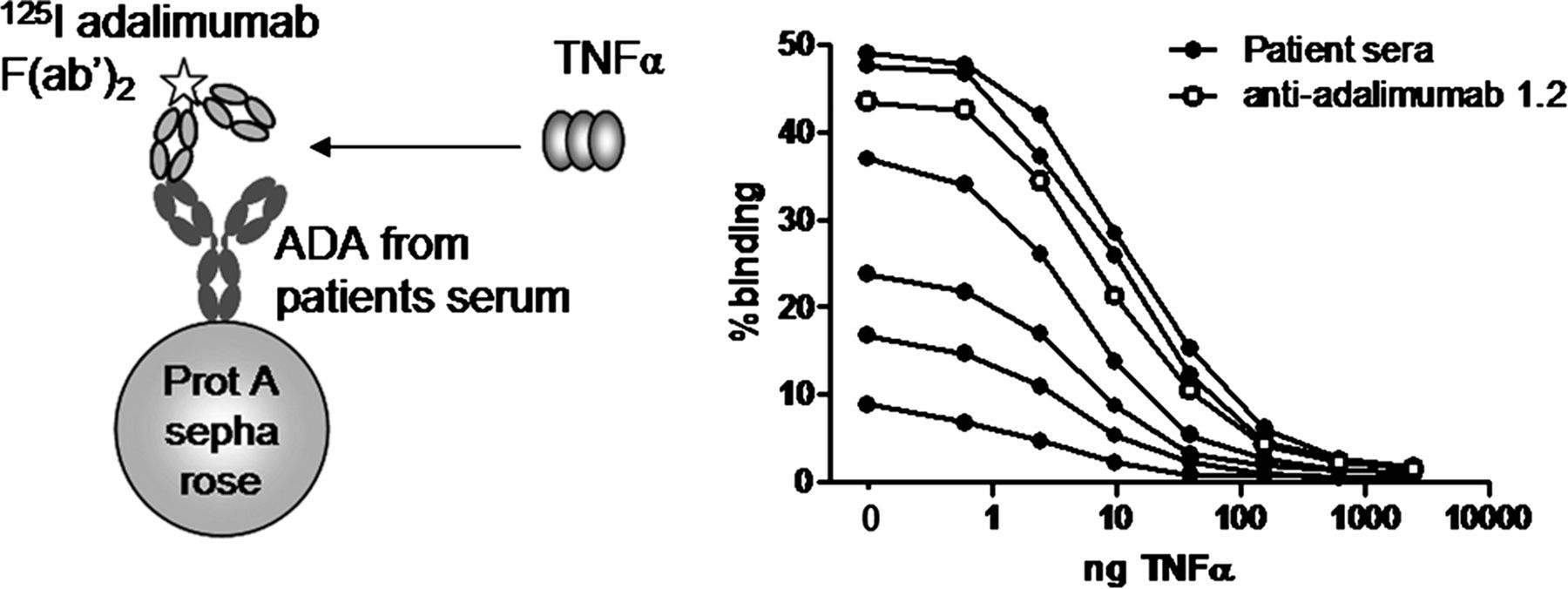
We next investigated whether anti-adalimumab 1.2 was directed against the TNFα-binding region of adalimumab using a TNFα bioassay (figure 3). In response to TNFα, ECRF-24 cells produce interleukin 8 (IL-8) which, as expected, can be inhibited by addition of adalimumab or infliximab (figure 3). Addition of an excess of anti-adalimumab 1.2 Fab fragments prevented inhibition of TNFα-induced IL-8 production by adalimumab, showing that anti-adalimumab 1.2 neutralises the effect of adalimumab while the inhibitory effect of infliximab was not affected (figure 3). In the absence of adalimumab, the recombinant monoclonal antibody had no agonistic or antagonistic effect on the ECRF-24 cells (see supplementary online figure 1).

The binding of adalimumab to tumour necrosis factor α (TNFα) can be neutralised by anti-adalimumab 1.2. In response to 1 ng TNFα, ECRF-24 cells produce interleukin 8 (IL-8) in the linear range of the titration curve. Both adalimumab and infliximab neutralise TNFα, thereby preventing IL-8 production (p<0.0001, adalimumab; p=0.0001, infliximab). Anti-adalimumab 1.2 rescues IL-8 production in the presence of adalimumab (p<0.0001), but not infliximab (n=3). Results represent mean and SEM (error bars). p Values are calculated using an unpaired t test.
All tested anti-adalimumab-positive patient serum samples were able to neutralise adalimumab activity in the TNFα bioassay, showing that in all patients at least part of the AAA are neutralising. To investigate which fraction of the polyclonal response is neutralising, we performed an ABT in which radiolabelled adalimumab F(ab′)2 was pre-incubated with increasing amounts of TNFα. The presence of AAA-drug complexes in serum may lead to false-positive results due to cross-linking of radiolabelled adalimumab F(ab′)2 to complex-derived adalimumab by the homotrimeric TNFα (see supplementary online figure 2). To circumvent this, we fractionated serum from six patients with high anti-adalimumab levels on sucrose gradients to separate the free monomeric AAA from AAA-drug complexes, as previously described.15 TNFα dose-dependently inhibited the binding to radiolabelled adalimumab F(ab′)2 of anti-adalimumab derived from serum from six patients as well as monoclonal anti-adalimumab 1.2 (figure 4). These data clearly show that the humoral immune response against adalimumab in all tested patients is restricted to the TNFα binding region of adalimumab.

Antibodies against adalimumab are neutralising antibodies. Binding of monomeric anti-adalimumab antibodies and anti-adalimumab 1.2 to radiolabelled adalimumab F(ab′)2 were significantly inhibited by increasing amounts of tumour necrosis factor α (TNFα) (p<0.005, paired t test).
The restricted immune response against adalimumab leads to small immune complexes
An antibody response against a very restricted immunogenic region, as we describe here for adalimumab, is expected to result in formation of small immune complexes which might not be efficiently cleared from the circulation. We recently described a new assay which is able to measure anti-drug antibodies in complex with adalimumab, enabling us to analyse the size of adalimumab/anti-adalimumab complexes in the serum of patients.15 AAA-positive patient serum samples with different levels of AAA and adalimumab collected just prior to the next administration of adalimumab were fractionated on sucrose gradients and the fractions containing adalimumab/anti-adalimumab complexes were measured. This revealed the presence of small immune complexes in all 14 patients tested. The results of three representative patients are shown in figure 5. In some patients, adalimumab/anti-adalimumab complexes were detected next to free uncomplexed anti-adalimumab while, in other patients, all AAA were present in small immune complexes. The measurement of marker proteins in the different fractions suggests that these complexes are no larger than IgG dimers. To aim for maximal separation of monomeric IgG and small immune complexes, sucrose gradients were run at 20°C. Under these conditions, larger immune complexes may end up at the bottom. However, additional experiments using sucrose gradients centrifuged at 4°C did not reveal any larger complexes (data not shown). Thus, the restricted antibody response against adalimumab leads to the formation of small circulating immune complexes which can be detected in patients receiving adalimumab therapy.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The restricted immune response against adalimumab leads to the formation of small immune complexes. Anti-adalimumab antibodies measured in sucrose gradient fractions of three representative patient serum samples. The measurements of marker proteins albumin (66 kD, fraction 17), IgG (150 kD, fraction 13) and C1q (410 kD, fraction 1) showed that the complexes were all the size of dimers. The top of the gradient is to the right of the figure.
Discussion
Antibody formation against adalimumab is increasingly being recognised as a major cause of treatment failure. However, the mechanism by which AAA affects treatment efficacy has not yet been elucidated. We propose two possible mechanisms. First, the formation of immune complexes between AAA and therapeutic antibody may lead to increased clearance of the drug resulting in suboptimal dosing of the drug. Second, the antibodies may be functionally neutralising, thus directly affecting treatment efficacy. This study shows that, in the case of adalimumab, virtually all antibodies against adalimumab are neutralising and the AAA response is very restricted, leading to the formation of small immune complexes. Small immune complexes are thought to clear slowly, suggesting that the effect of AAA formation on treatment efficacy is probably mainly dependent on neutralisation of the drug. This implies that, for adalimumab, routine measurements of binding antibodies will be sufficient without requirement to test for their neutralising capacity. In the light of these data, it would be very valuable to determine if the antibody response against human therapeutic antibodies is always restricted to the idiotype and to analyse if the immune response against humanised and chimeric monoclonal antibodies is less restricted.
In view of this new finding that all antibodies are neutralising, it might seem contradictory that only some AAA-producing patients lose responsiveness. However, we hypothesise that the clinical response is dependent on the magnitude of the immune response. Low AAA levels might not alter the clinical response of patients since only a small portion of the adalimumab is neutralised. These patients would therefore still have sufficient drug levels to respond to treatment. Only in patients who produce high levels of ADA is most or all of the adalimumab neutralised leading to non-response to treatment.
Another mechanism by which AAA could interfere with the effectiveness of the therapeutic monoclonal antibody is in cases where AAA bear an ‘internal image’ of the antigen. Previous studies have shown that an antigen bound by a particular antigen combining region (idiotype) of an antibody may be functionally mimicked by a secondary anti-idiotype antibody that binds to the same region of that idiotype. For therapeutic antibodies, this carries the risk that anti-idiotype antibodies against the therapeutic antibody may resemble the target protein and, in the case of adalimumab, therefore react with the TNF receptor. This could be detrimental for the effectiveness of the therapeutic monoclonal antibody. However, the anti-idiotype monoclonal antibodies against adalimumab that we isolated and subsequently cloned showed no effect on the TNF receptor tested in a TNFα bio-assay (see supplementary online figure 1). Reactivity of anti-idiotype antibodies against adalimumab with the TNF receptor can only be expected if adalimumab binds to exactly the same region on TNFα as the TNF receptor.
Knowledge of the mechanism by which AAA interfere with drug efficacy can be important for clinical purposes. For instance, the restrictive nature of the response against adalimumab can be explained by either one major immunogenic idiotope in adalimumab or steric hindrance when all antibodies are binding to idiotopes in close proximity to each other on the idiotype of adalimumab. It would be interesting to investigate this further and identify the precise immunogenic B cell epitopes of adalimumab. Eventually this could lead to the production of less immunogenic variants of adalimumab. Of course, alterations may subsequently lead to the introduction of new immunogenic epitopes or loss of function of the therapeutic antibody.
The presence of large amounts of small immune complexes may have clinical consequences. On the one hand it might be expected that the clinical consequences of small immune complexes are limited because of little complement activation and FcγR triggering. On the other hand, the existence of long lasting small immune complexes may increase the risk of type III hypersensitivity reactions such as those occurring in diseases like serum sickness and systemic lupus erythematosus. A recently published paper showed an increased incidence of severe venous and arterial thromboembolic events in patients who produce AAA against adalimumab.18 It would be interesting to investigate whether the presence of these immune complexes leads to serious side effects. In this light, our previous finding that most patients make antibodies against adalimumab, even at levels that do not preclude full neutralisation, gains new importance.15 It is likely that, in most patients, small circulating immune complexes are chronically induced.
In conclusion, the following picture emerges. After repeated treatment with adalimumab most patients develop AAA. The antibodies are hypermutated and of the IgG1 and IgG4 isotype. The antibody response is very restricted to the idiotype region of adalimumab. All antibodies neutralise adalimumab function and give rise to small immune complexes which can be detected in the circulation of patients being treated with the therapeutic antibody. Depending on the magnitude of the immune response, the clinical effect might be limited if only a small portion of the adalimumab is neutralised. However, if high levels of AAA are produced, all the adalimumab is neutralised leading to clinical non-response.
Acknowledgments
The authors thank Eric Mul and Floris van Alphen for their help with the FACS sorting, Gestur Vidarsson for sharing his expertise on the different cloning steps and Paul Parren for critically reading the manuscript.
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online Methods
- Data supplement 2 - Online Figure 1
- Data supplement 3 - Online Figure 2
Footnotes
-
Contributors Study concept and design: PAvS, RNdJ, SMvH, TR, LA, GJW, DW. Acquisition of data: PAvS, LAvdS, RNdJ, EELvB, SK, EdG, MH. Analysis and interpretation of data: PAvS, LAvdS, EELvB, SMvH, TR, LA, GW, DW. Clinical revision of the manuscript for important intellectual content: PAvS, LAvdS, RNdJ, SMvH, TR, LA, GW, DW. Obtained funding: GW. Study supervision: LA, GW, DW.
-
Funding Funding of this study was provided by an unrestricted grant of Wyeth Pharmaceuticals. Wyeth Pharmaceuticals had no involvement in the study design; in the collection, analysis, and interpretation of data; in the writing of the manuscript; or in the decision to submit the manuscript for publication.
-
Competing interests GJW has received a research grant from Wyeth Pharmaceuticals and honoraria for lectures from Amgen and Pfizer. LA has received honoraria for lectures from Abbott, Roche and Pfizer.
-
Patient consent None.
-
Ethics approval Ethics approval was obtained from the medical ethics committee of the Jan van Breemen Institute, Amsterdam.
-
Provenance and peer review Not commissioned; externally peer reviewed.