Article Text

Abstract
Objectives Pathologic fibroblast activation drives fibrosis of the skin and internal organs in patients with systemic sclerosis (SSc). β-catenin is an integral part of adherens junctions and a central component of canonical Wnt signaling. Here, the authors addressed the role of β-catenin in fibroblasts for the development of SSc dermal fibrosis.
Methods Nuclear accumulation of β-catenin in fibroblasts was assessed by triple staining for β-catenin, prolyl-4-hydroxylase-β and 4′,6-diamidino-2-phenylindole (DAPI). The expression of Wnt proteins in the skin was analysed by real-time PCR and immunohistochemistry. Mice with fibroblast-specific stabilisation or fibroblast-specific depletion were used to evaluate the role of β-catenin in fibrosis.
Results The auhors found significantly increased nuclear levels of β-catenin in fibroblasts in SSc skin compared to fibroblasts in the skin of healthy individuals. The accumulation of β-catenin resulted from increased expression of Wnt-1 and Wnt-10b in SSc. The authors further showed that the nuclear accumulation of β-catenin has direct implications for the development of fibrosis: Mice with fibroblast-specific stabilisation of β-catenin rapidly developed fibrosis within 2 weeks with dermal thickening, accumulation of collagen and differentiation of resting fibroblasts into myofibroblasts. By contrast, fibroblast-specific deletion of β-catenin significantly reduced bleomycin-induced dermal fibrosis.
Conclusions The present study findings identify β-catenin as a key player of fibroblast activation and tissue fibrosis in SSc. Although further translational studies are necessary to test the efficacy and tolerability of β-catenin/Wnt inhibition in SSc, the present findings may have clinical implications, because selective inhibitors of β-catenin/Wnt signaling have recently entered clinical trials.
Statistics from Altmetric.com
Introduction
Fibrosis of the skin and internal organs is a key feature of systemic sclerosis (SSc).1 Since fibrosis can disrupt the physiological tissue architecture and lead to organ failure, it causes much of the morbidity and mortality in patients with SSc.2 Fibrosis arises from excessive accumulation of extracellular matrix (ECM) components released by pathologically activated fibroblasts.3 So far, the molecular mechanisms that underlie the aberrant fibroblast activation with persistent expression of contractile proteins and exorbitant release of ECM components are incompletely understood.
β-catenin has a dual role in cells. It is an integral part of adherens junctions and acts as a cytoplasmic adapter molecule to anchor cadherins (transmembrane proteins that establish the junctions with neighboring cells) to the actin cytoskeleton. Thus, β-catenin stabilises cell-cell-adhesions, which is essential for normal cell physiology and tissue architecture.4 In addition to its structural role, β-catenin operates as a transcriptional co-activator of the T cell factor (TCF) family of DNA-binding proteins. This links β-catenin to canonical Wnt signaling, in which β-catenin processes signals from various Wnts to modulate gene transcription.5 In the absence of Wnt signals, a so-called ‘destruction complex’ comprising of adenomatosis polyposis coli, axin, glycogen synthase kinase-3β (GSK-3β), and casein kinase phosphorylates β-catenin, which promotes subsequent degradation of β-catenin.6 Binding of Wnts to their receptors, however, disrupts the ‘destruction complex’, and thus prevents degradation of β-catenin. Unphosphorylated β-catenin accumulates and translocates to the nucleus, where it binds to the family of TCF proteins and stimulates the transcription of target genes, such as axin-2.7 Whereas physiologic β-catenin/Wnt signaling is crucial for normal organ development and tissue homeostasis, dysregulation of this pathway plays a central role in various diseases, including cancer, arthritis and osteoporosis.8,–,11 Accumulating evidence further indicates that enhanced canonical Wnt signaling might play an important role in fibrotic diseases, including pulmonary and renal fibrosis as well as hypertrophic scars.12,–,17 Of particular interest, several members of the Wnt pathway appear to be up-regulated in animal models of SSc and in fibrotic human skin, indicating that Wnt signaling might regulate fibroblast activation in SSc.18 19
Our study aimed to establish the role of β-catenin for tissue fibrosis in SSc. We demonstrated that increased expression of Wnt proteins in SSc patients leads to nuclear accumulation of β-catenin in SSc fibroblasts. We could further show that this accumulation of β-catenin has profound effects on fibroblast activation. In in vivo-models, fibroblast-specific stabilisation of β-catenin results in increased release of collagen and prominent dermal fibrosis, whereas fibroblast-specific deletion of β-catenin inhibits experimental fibrosis.
Material and methods
Patients
Skin biopsies were obtained from involved skin at the volar aspect of the forearm of 18 patients with SSc. All patients fulfilled the criteria for SSc as defined by LeRoy et al.20 The study included 13 female and 5 male patients. The median age was 51 years, ranging from 20 to 71, and median disease duration 6 years, ranging from 1 to 13 years. Seven patients suffered from limited cutaneous disease, 11 from the diffuse disease subtype. Prior to biopsy, patients have not received any disease-modifying anti-rheumatic drug treatment. Age- and sex-matched healthy volunteers served as controls.
Immunofluorescence staining for prolyl-4-hydroxylase-β and β-catenin
Formalin-fixed, paraffin-embedded skin sections from healthy individuals and SSc patients were stained with antiprolyl-4-hydroxylase-β (Acris Antibodies GmbH, Herford, Germany) and anti-β-catenin (R&D Systems, Ambington, UK). Concentration-matched and species-specific immunoglobulins (Vector Laboratories, Burlingame, California, USA) were used as control antibodies. After labeling with rhodamine-tagged (red, prolyl-4-hydroxylase-β) and Alexa Fluor 488-tagged (green, β-catenin; both molecular probes) secondary antibodies, and staining of nucleic acids with DAPI, slices were analysed at 200- and 1000-fold magnification.
Immunohistochemistry for Wnt-1, Wnt-4 and Wnt-10b
Skin sections from healthy individuals and SSc patients were stained with anti-Wnt-1 (Abcam, Cambridge, UK), anti-Wnt-4 (Abgent, San Diego, California, USA) and anti-Wnt-10b antibodies (ProSci incorporated, Poway, California, USA). Concentration-matched and species-specific immunoglobulins (Vector Laboratories, Burlingame, California, USA) served as control antibodies. After incubation with polyvalent secondary antibody KIT (IDLabs, London, Ontario, Canada) and 3,3-diaminobenzidine (DAB) peroxidase substrate solution, slices were analysed at 200-fold magnification.
Quantitative real time-PCR
Messenger RNA was isolated using the NucleoSpin RNA II Kit (Machery Nagel, Düren, Germany). After reverse transcription, gene expression was quantified by SYBR Green real time-PCR using the ABI Prism 7300 Sequence Detection System (Applied Biosystems, Foster City, California, USA). Specific primer pairs are listed in table 1. β-actin was used for normalisation of samples. Samples without enzyme in the reverse transcription reaction (non-RT controls) were used as negative controls. Unspecific signals caused by primer dimers were excluded by non-template controls and by dissociation curve analysis.
Primers of Wnt signaling
Fibroblast-specific over-expression and deletion of β-catenin in vivo
To selectively modify the expression and stability of β-catenin in fibroblasts in vivo, we used the Col1a2; Cre-ER mouse model. In Col1a2; Cre-ER mice, a fibroblast-specific enhancer of the mouse Col1a2 gene directs the expression of the polypeptide consisting of a fusion between Cre recombinase and a mutant ligand-binding domain of the estrogen receptor. In these mice, administration of tamoxifen allows the Cre recombinase to enter the nucleus in fibroblastic cells.21 22 To generate mice with fibroblast-specific stabilisation of β-catenin, Col1a2; Cre-ER mice were cross-bred with ΔExon 3 β-cateninfl/fl mice with loxP sites flanking the exon 3 of the β-catenin gene. For mice with fibroblast-specific deletion of β-catenin, Col1a2; Cre-ER mice were cross-bred with Ctnnb1fl/fl mice carrying the loxP sites in introns 1 and 6 of the β-catenin gene. Thus, we generated mice with the following two genotypes: (i) Mice that expressed the Col1a2; Cre-ER mutation and were heterozygous for the ΔExon 3 β-catenin mutation (ΔExon 3 β-cateninfl/wt x Col1a2; Cre-ER; abbreviation: ΔEx3), and (ii) mice carrying the Col1a2; Cre-ER mutation, which were homozygous for the Ctnnb1 mutation (Ctnnb1fl/fl x Col1a2; Cre-ER; abbreviation: Ctnnb1). We back-crossed all mice on a C57BL/6 background for more than 20 generations. Col1a2; Cre-ER mice were kindly provided by de Crombrugghe (MD Anderson Cancer Center, Houston, Texas, USA), ΔExon 3 β-cateninfl/fl were kindly provided by M M Maketo (Kyoto University, Kyoto, Japan), and Ctnnb1fl/fl mice were purchased from Jackson Laboratories (Bar Harbor, Maine, USA).
In ΔEx3 mice, loss of exon 3 led to intracellular stabilisation and accumulation of β-catenin, since the exon 3 encodes for important serine/threonine residues, which are targets of phosphorylation and subsequent degradation of β-catenin.23 Cre was activated by intraperitoneal injections of tamoxifen at a dose of 1 mg/d for 5 subsequent days at the age of 4 weeks. Control animals were treated with corn oil, the solvent of tamoxifen. Mice were sacrificed at 2, 4 and 8 weeks after Cre activation.
To induce a fibroblast-specific deletion of β-catenin, 4-week-old, sex-matched Ctnnb1 mice were injected intraperitoneally with tamoxifen according to the above-mentioned protocol. At the age of 6 weeks, we challenged mice with local injections of bleomycin for 4 weeks as described previously.21 24 25 For both mouse models, dermal fibrosis was quantified by measuring dermal thickness, hydroxyproline content and the number of myofibroblasts as described previously.26,–,28 The local ethical committee approved all animal experiments.
Statistics
All data are presented as median with IQR, and differences between the groups were tested for their statistical significance by non-parametric Mann–Whitney U test. A p value of less than 0.05 was considered statistically significant; p values are expressed as follows: n.s for not significant; 0.05>p>0.01 as*; 0.01>p>0.001 as **; p<0.001 as ***.
Results
Nuclear accumulation of β-catenin in dermal SSc fibroblasts
To examine the role of β-catenin for fibroblast activation in SSc, we first determined the subcellular localisation of β-catenin in fibroblasts. Triple-staining for β-catenin, the fibroblast-specific marker prolyl-4-hydroxylase-β, and the chromatin marker DAPI showed increased nuclear accumulation of β-catenin in SSc fibroblasts compared to healthy volunteers (figure 1A, B). Nuclear staining for β-catenin was observed in 82.9% of fibroblasts in SSc skin but only in 27.9% of dermal fibroblasts in normal skin (p=0.001).

β-catenin is stabilised in fibroblasts in SSc skin. (A) Immunofluorescence images show staining against chromatin (blue), β-catenin (green), and prolyl-4-hydroxylase-β (red) as well as overlay of these three stains in skin sections from patients with SSc and healthy individuals. Images are in 200- and 1000-fold magnifications. (B) Quantification of nuclei that stained for β-catenin, DAPI and prolyl-4-hydroxylase divided by the total number of DAPI and prolyl-4-hydroxylase-positive fibroblastic cells. Values are expressed in per cent of β-catenin-positive nuclei.
To identify potential Wnt ligands that induce stabilisation and nuclear translocation of β-catenin in SSc, we measured expression profiles of all canonical Wnt proteins in the skin. Wnt-2b-13, Wnt-3a, Wnt-6, Wnt-7a, Wnt-7b and Wnt-8a were not detectable in any sample by real-time PCR. We detected mRNA for Wnt-2, Wnt-9a, Wnt-9b and Wnt-16 in only few samples without differences between SSc patients and controls. By contrast, we found Wnt-1 and Wnt-4 expressed in the majority of SSc samples with trends towards increased expression in SSc (p=0.128 and p=0.034). Wnt-10b mRNA was expressed in all SSc biopsies, but in none of the controls (p=0.002). We detected Wnt-10a mRNA in most SSc samples but not in control biopsies. The expression levels, however, were rather low compared to Wnt-1, Wnt-4 and Wnt-10b (figure 2A). To confirm potential differences in the expression on protein levels, we performed immunohistochemistry for Wnt-1, Wnt-4 and Wnt-10b (figure 2B). We did not observe any differences in the intensity or staining pattern for Wnt-4. The expression of Wnt-1 and Wnt-10b, however, were strongly increased in the skin from SSc patients compared to healthy individuals. In fibrotic SSc skin from most of the patients, we found a strong staining for those two Wnt ligands in fibroblasts as well as blood vessels and the epidermis. By contrast, Wnt-1 and Wnt-10b were barely detectable in non-fibrotic healthy skin (figure 2B).
Consistent with increased activation of canonical β-catenin/Wnt signaling, we also observed increased mRNA levels of axin-2 in SSc skin, a classical target gene of β-catenin (figure 2C). When we finally compared samples from the two disease subtypes (diffuse vs limited), we did not find significant differences in either β-catenin levels or Wnt expression or axin-2 levels.

Stabilisation of β-catenin in dermal fibroblasts is induced by increased expression of canonical Wnt ligands. (A) mRNA expression profiles of all canonical Wnt ligands in skin from patients with SSc and healthy individuals. mRNA levels of the Wnt ligands are illustrated in relation to the corresponding β-actin values (internal control). Percentage of skin samples that expressed specific Wnt ligands are illustrated below the graph. (B) Immunohistochemistry demonstrated increased expression of Wnt-1 and Wnt-10b but not of Wnt-4 in skin sections of SSc patients. Representative images are shown at 200-fold magnification. (C) Increased mRNA levels of axin-2 in the skin of SSc patients. The relative expression of axin-2 in the SSc samples compared to control samples is expressed as x-fold increase.
Fibroblast-specific stabilisation of β-catenin causes spontaneous experimental skin fibrosis
To further analyse whether the nuclear accumulation of β-catenin in SSc fibroblasts contributes to fibroblast activation and tissue fibrosis, we created mice with fibroblast-specific, tamoxifen-inducible stabilisation of β-catenin (ΔEx3 mice). ΔEx3 mice developed progressive dermal fibrosis even within 2 weeks after Cre activation with a significant increase in skin thickness by 61.3% (p=0.002) compared to control littermates (figure 3A, B). Skin thickening further progressed at 4 weeks and culminated in an incline of 102.6% (p<0.001) after 8 weeks. These findings were in line with a strong increase in the dermal hydroxyproline content (figure 3C). Two weeks after Cre activation, the hydroxyproline content rose by 108.5% (p=0.037) and it progressed to 327.8% (p=0.002) after 8 weeks (figure 3C). We also observed significantly higher numbers of myofibroblasts with maximal increases of 408.8% (p=0.004) at 4 weeks after Cre activation (figure 3D). In contrast to the dramatic pathologies in the skin, mice did not show significant collagen accumulation neither in the lungs nor the gastrointestinal tract during the observation period (data not shown).
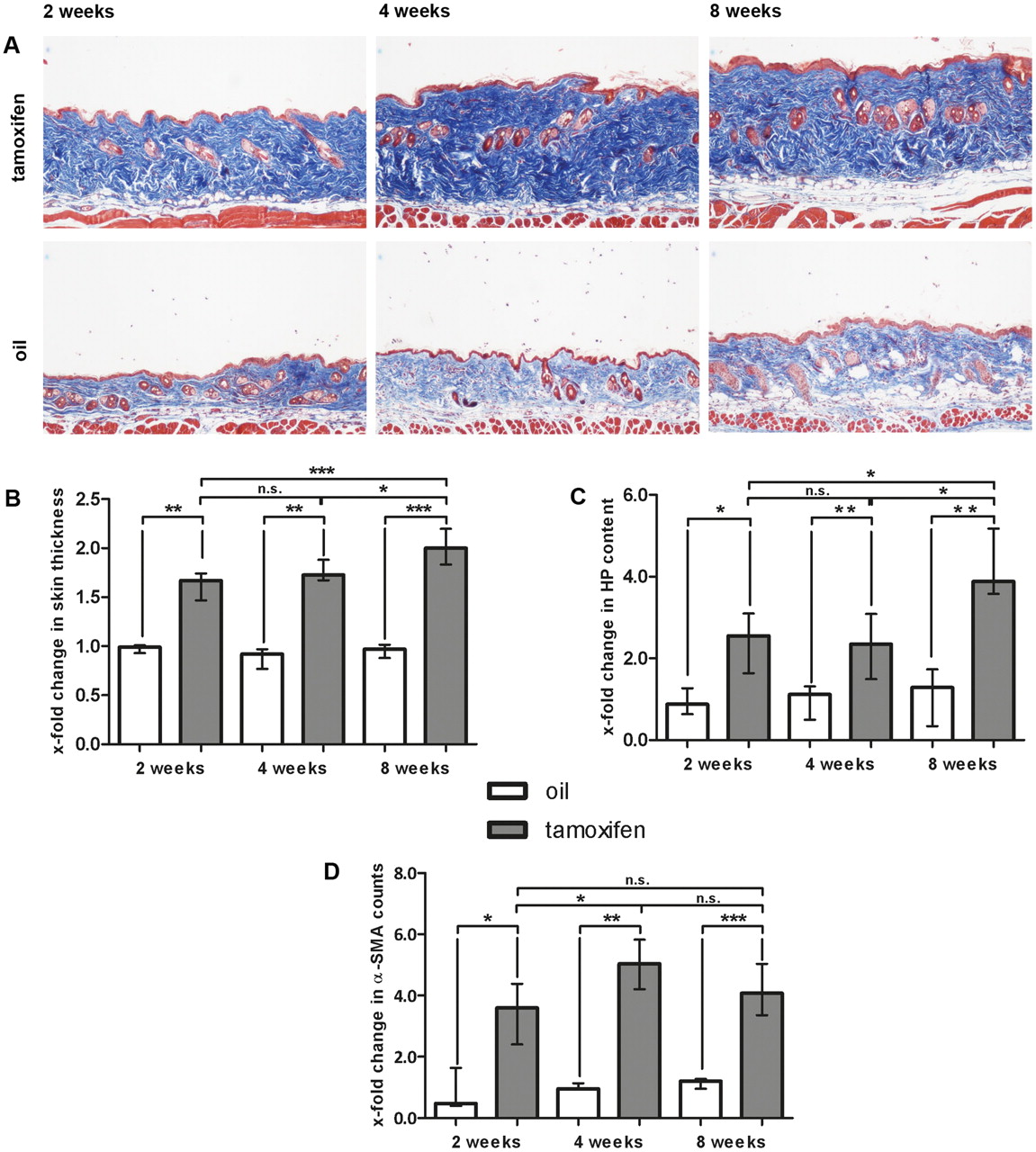
Development of progressive dermal fibrosis in ΔEx3 mice with fibroblast-specific stabilisation of β-catenin. Cre-mediated recombination resulting in fibroblast-specific activation of β-catenin was induced by tamoxifen injection (n=10). Mice of the same genotype injected with oil served as controls (n=7). (A) Trichrome staining with blue staining for collagens. Pictures are shown at 100-fold magnification. (B) Skin thickening as determined by H and E staining. Values are expressed in relation to skin thickness of non-activated, oil-treated mice 2 weeks after mock treatment. White bars show non-activated, oil-treated ΔEx3 mice and grey bars represent skin thickness of activated, tamoxifen-treated ΔEx3 mice. (C) Hydroxyproline content in non-activated (white bars) and activated (grey bars) ΔEx3 mice. Values are expressed in relation to hydroxyproline content of non-activated, oil-treated mice at week 2. (D) α-SMA positive myofibroblasts in the skin from non-activated (white bars) and activated (grey bars) ΔEx3 mice. Values are expressed in relation to α-SMA counts of non-activated, oil-treated mice 2 weeks after Cre activation.
Fibroblast-specific deletion of β-catenin prevents experimental dermal fibrosis
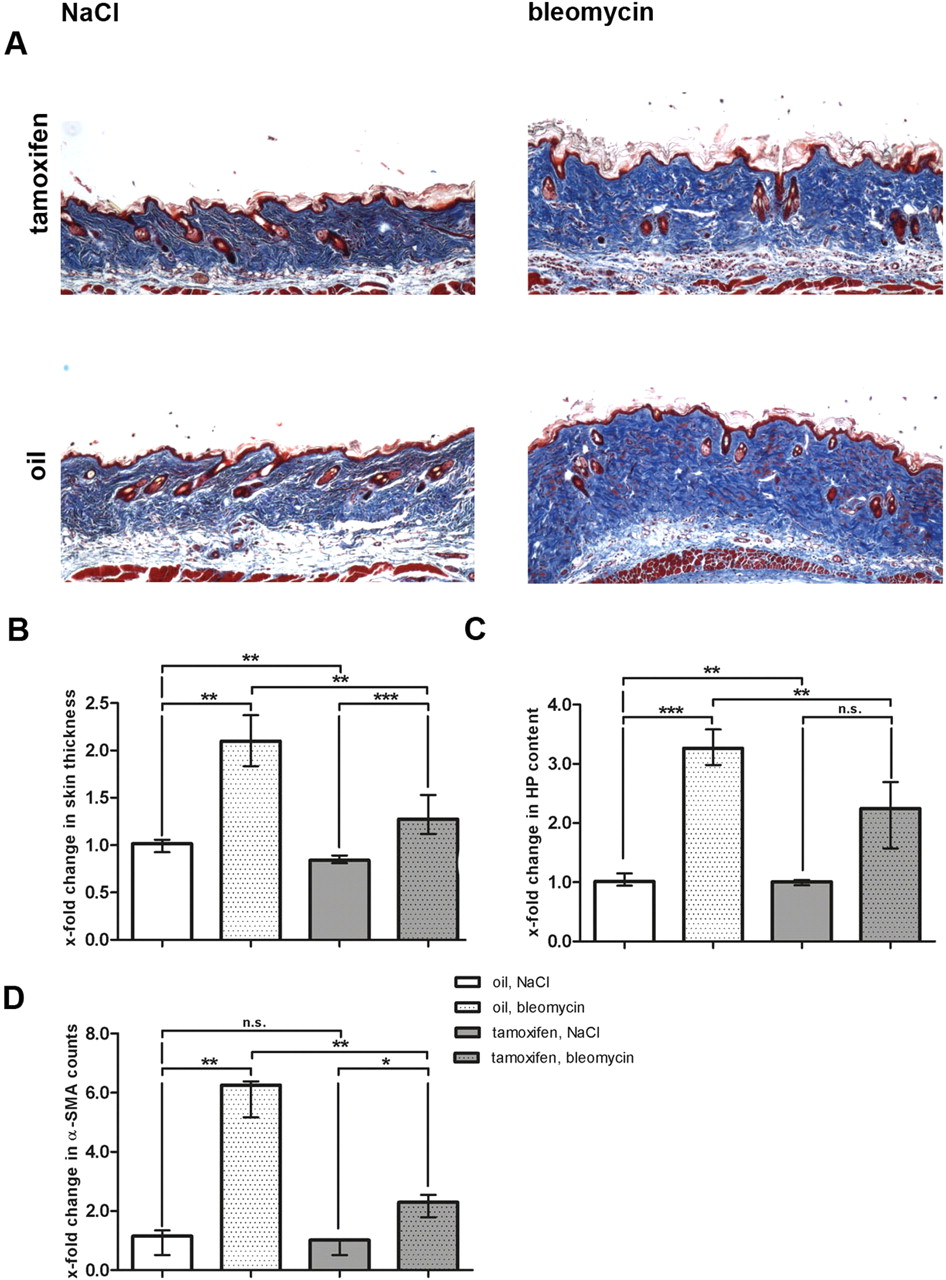
After having shown that stabilisation of β-catenin is sufficient to induce fibrosis, we investigated whether fibroblast-specific deletion of β-catenin inhibits fibrosis. We observed that bleomycin-induced dermal fibrosis was significantly reduced in Cre-activated Ctnnb1 mice (figure 4). In oil-treated, non-activated littermates with normal levels of β-catenin, the dermal thickness increased by 110.3% (p=0.001) after bleomycin challenge (figure 4A, B). By contrast, in mice with fibroblast-specific deletion of β-catenin, the dermal thickness increased by only 31.9%. Thus, fibroblast-specific deletion of β-catenin reduced dermal thickening upon challenge with bleomycin by 71.1% (p=0.004) (figure 4B). In line with these results, the hydroxyproline content was decreased by 49.7% (p=0.001; figure 4C). Deletion of β-catenin also prevented myofibroblast differentiation with reduction in myofibroblast counts by 75.4% (p=0.006; figure 4D). Thus, fibroblast-specific deletion of β-catenin efficiently prevented bleomycin-induced dermal fibrosis confirming a key role of β-catenin in fibroblast activation.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Prevention of bleomycin-induced experimental dermal fibrosis in Ctnnb1 mice with fibroblast-specific depletion of β-catenin. Cre-mediated recombination was activated by tamoxifen injection. Mice of the same genotype injected with oil were used as controls. For all groups n=6. (A) Trichrome staining with blue staining for collagens (100-fold magnification). (B) Skin thickening as determined by H and E staining. Values are expressed in relation to skin thickness of non-activated (oil), NaCl-treated mice. (C) Hydroxyproline content expressed in relation to hydroxyproline content of non-activated (oil), NaCl-treated mice. (D) α-SMA positive myofibroblasts expressed in relation to α-SMA counts in non-activated (oil), NaCl-treated mice.
Discussion
We demonstrate in the present study that β-catenin accumulates in SSc fibroblasts. Nuclear staining for β-catenin was observed in most fibroblasts in skin sections of SSc patients, but only in few fibroblasts in normal skin. The stabilisation of β-catenin in SSc fibroblasts resulted from increased expression of Wnt proteins. Screening of all members of canonical Wnt ligands highlighted that Wnt-1 and in particular Wnt-10b were overexpressed in SSc skin. Furthermore, stabilisation of β-catenin resulted in the increased transcription of the β-catenin target gene axin-2. Consistent with our findings, increased mRNA levels of Wnt-2 and Wnt-10b have been described in tight-skin-1 mice and in bleomycin-induced dermal fibrosis, respectively, two common animal models of SSc.18 19 The nuclear accumulation of β-catenin in fibroblasts may not be restricted to dermal fibrosis, but may also occur in pulmonary fibrosis.29
Given the prominent nuclear accumulation of β-catenin in SSc fibroblasts and the crucial role of β-catenin for tissue homeostasis, we hypothesised that the stabilisation of β-catenin in SSc fibroblasts might have direct implications for tissue fibrosis. In this context, indirect evidence comes from studies on the role of glycogen synthase kinase-3. Inhibition of glycogen synthase kinase-3β (GSK-3β) activated canonical Wnt signalling and exacerbated experimental tissue fibrosis30 In the current study, we specifically targeted β-catenin and indeed found that fibroblast-specific stabilisation of β-catenin resulted in rapid and progressive fibrosis with prominent dermal thickening, differentiation of resting fibroblasts into myofibroblasts and increased accumulation of collagens. Of note, prominent fibrosis was already evident as early as 2 weeks after activation of Cre recombinase, highlighting the potent pro-fibrotic effects of β-catenin in fibroblasts. A previous study by Cheon et al. showed that ubiquitous expression of a mutated human β-catenin/c-myc construct resulted in aggressive fibromatosis within 3 months.31 In the present study, we observed the development of massive fibrosis, but none of our mice developed fibromas even after prolonged observation for 6 months. Several differences in the mouse models used in the two studies might account for the different phenotypes: (i) We used a 6-kilobase transcriptional enhancer from the far-upstream region of the mouse proα2(I)collagen gene to stabilise β-catenin selectively in fibroblasts. By contrast, the expression of the β-catenin construct in the study by Cheon and coworkers was not restricted to fibroblasts but was ubiquitous. Given the cell-type dependent effects of β-catenin, activation of β-catenin/Wnt signaling in other cell types might have contributed to the formation of fibromas, e.g. by providing additional growth stimulation.32 (ii) In contrast to our study, Cheon et al. co-expressed myc as a tag together with β-catenin. Myc is a prototypical oncogene and aberrant expression of myc has been implicated into a broad spectrum of different neoplasias. In fibroblasts, myc induces uncontrolled proliferation and transformation.33 Thus, the myc overexpression is likely to contribute to the development of fibromas. (iii) In addition to the myc tag, other differences in the expression constructs might also have led to the different outcomes. Cheon and coworkers used a human β-catenin transgene with mutations at codons S33, S37, T41 and S45, whereas our experiments based on a murine transgene with deletion of the complete exon 3 encoding all of these residues.23
The highly increased nuclear levels of β-catenin together with its potent pro-fibrotic effects suggest that β-catenin might be a candidate for anti-fibrotic approaches. Indeed, we could show that fibroblast-specific depletion of β-catenin prevents bleomycin-induced dermal fibrosis. Dermal thickening, myofibroblast differentiation and hydroxyproline content were significantly reduced in activated Ctnnb1 mice compared to non-activated control mice. Our findings may have translational implications, but additional preclinical studies will have to address the efficacy of β-catenin/Wnt signaling inhibition in the treatment of pre-established fibrosis as well as to investigate the most suitable targets for pharmacologic therapy within the β-catenin/Wnt pathway.
Although targeting β-catenin/Wnt signaling has been challenging in the past, promising approaches including porcupine inhibitors, drugs that prevent the interaction between β-catenin and TCF transcription factors, and tankyrase inhibitors are emerging.34 Some of those, such as CWP232291, which promotes the degradation of β-catenin, are currently evaluated in clinical trials (www.clinicaltrials.gov) and would be available for SSc patients. Nevertheless, given its essential role in stem cell maintenance, non-selective inhibition of β-catenin/Wnt signaling is likely to be limited by adverse effects. For example, Wnt signaling is crucial for regeneration of intestinal mucosa, and thus, global inhibition of Wnt signaling is predicted to cause intestinal toxicity.34 This limitation might be circumvented by tissue or cell-specific approaches. In this context, we showed that fibroblast specific depletion of β-catenin in Ctnnb1 mice did not result in clinically obvious pathological changes with normal activity, body weight, food intake, and behavior. Macroscopic examination of the colon as a tissue that would be particularly sensitive to Wnt inhibition did not reveal abnormalities. Thus, fibroblast-specific blockade of β-catenin/Wnt signaling might efficiently reduce fibrosis in patients with SSc while showing a favorable safety profile.
In summary, we demonstrated that overexpression of Wnt proteins leads to accumulation of β-catenin in SSc fibroblasts. Selective stabilisation of β-catenin in fibroblasts results in excessive release of ECM and rapid development of dermal fibrosis. In contrast, fibroblast-specific depletion of β-catenin protects from experimental fibrosis. Thus, our findings identified β-catenin as a key player of fibroblast activation and tissue fibrosis in SSc.
Acknowledgments
The authors thank Maria Halter and Anna-Maria Herrmann for excellent technical assistance.
References
Footnotes
-
Funding Christian Beyer: Research scholar at the Interdisciplinary Center of Clinical Research (IZKF) in Erlangen; Grant from the Erlanger Leistungsbezogene Anschubfinanzierung und Nachwuchsförderung (ELAN). Jörg Distler: Grant A40 of the Interdisciplinary Center of Clinical Research (IZKF) in Erlangen; grants from the Deutsche Forschungsgesellschaft; and the Career Support Award of Medicine of the Ernst Jung Foundation.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.