Article Text

Abstract
Objectives The hallmark of systemic sclerosis (SSc) is the accumulation of extracellular matrix proteins by pathologically activated fibroblasts. This study analysed the antifibrotic effects of the selective c-Jun N-terminal kinase (JNK) inhibitor, CC-930, which recently entered first clinical trials as a novel antifibrotic approach.
Methods Phosphorylated c-Jun was detected by western blot and immunohistochemistry. The model of bleomycin-induced dermal fibrosis and the tight skin 1 (TSK1) mouse model were used to investigate the effects of CC-930 on the prevention of experimental fibrosis. The potential of CC-930 to induce regression of fibrosis was assessed in a modified model of established fibrosis.
Results Transforming growth factor beta (TGFβ) and platelet-derived growth factor (PDGF) activate JNK and stimulate the phosphorylation of its downstream target c-Jun. Incubation with CC-930 prevented the phosphorylation of c-Jun and reduced the stimulatory levels of these cytokines on the release of collagen. Inhibition of JNK prevented dermal thickening, myofibroblast differentiation and the accumulation of collagen in a dose-dependent manner in mice challenged with bleomycin and in TSK1 mice. In addition to the prevention of fibrosis, treatment with pharmacologically relevant doses of CC-930 also induced regression of established experimental fibrosis.
Conclusions These data identify JNK as a downstream mediator of the pro-fibrotic effects of of TGFβ and PDGF in SSc fibroblasts. Selective inhibition of JNK by CC-930 exerted potent antifibrotic effects in vitro and in different models in vivo. JNK might thus be a novel molecular target for the treatment of fibrosis in SSc.
Statistics from Altmetric.com
Systemic sclerosis (SSc) is a connective tissue disease that affects the skin and a variety of internal organs including lungs, gastrointestinal tract and heart. The predominant changes in the early stages of SSc are perivascular inflammatory infiltrates and apoptosis of microvascular endothelial cells. Later stages of SSc are characterised by massive accumulation of extracellular matrix components such as different types of collagen and fibronectin leading to progressive tissue fibrosis.1 The accumulating extracellular matrix disrupts the physiological tissue structure and frequently results in dysfunction of the affected organs. Tissue fibrosis is a major cause of morbidity and a major contributor to the increased mortality in SSc.2 3 The accumulation of extracellular matrix components in SSc is caused by an increased production of extracellular matrix by activated fibroblasts.4 Profibrotic cytokines such as transforming growth factor beta (TGFβ) and platelet-derived growth factor (PDGF) are important players in the pathogenesis of SSc as they potently activate fibroblasts and are sufficient to induce fibrosis in vivo.5 However, the intracellular signalling cascades, by which these cytokines stimulate the production of extracellular matrix and induce fibrosis are incompletely understood.
Jun N-terminal kinases (JNK) belong to the superfamily of mitogen-activated protein kinases (MAPK). JNK are phosphorylated and thereby activated in response to extracellular stimuli such as cytokines.6 JNK convert these extracellular stimuli into intracellular responses by activating downstream target genes such as members of the activator protein 1 (AP-1) family of transcription factors. JNK control cellular activation, differentiation and proliferation and thereby play key roles in the regulation of tissue homeostasis and inflammation.7 Indirect evidence suggests that JNK might also play a role in fibrotic diseases: JNK might be activated by pro-fibrotic cytokines. TGFβ, which potently stimulates the activation of resting fibroblasts and induces the release of extracellular matrix, activates JNK in epithelial cells.8 9 Target genes of JNK, such as the AP-1 family members c-Jun and other members of the AP-1 family have been implicated in the pathogenesis of fibrosis in SSc.10,–,14 However, the roles of JNK in SSc and potential therapeutic effects of JNK inhibitors have not been evaluated. Antifibrotic effects of JNK inhibitors in animal models of SSc would have direct translational implications because CC-930 is currently under evaluation in the clinic (www.clinicaltrials.gov; trial identifier NCT01203943). The first results indicate that JNK can be selectively targeted in humans by CC-930,15 and that inhibition of JNK is well tolerated with a low rate of severe adverse events.15 16
The aim of the present study was to define the role of JNK in fibroblast activation and tissue fibrosis in SSc and to evaluate the therapeutic potential of the selective inhibition of JNK. We demonstrate that JNK is activated by pro-fibrotic cytokines in SSc fibroblasts. Inhibition of JNK by the selective inhibitor CC-930 inhibits the release of extracellular matrix proteins in cultured fibroblasts, prevents fibrosis in different models of SSc, and induces regression of pre-established fibrosis without toxic side effects. JNK might thus be a novel molecular target for the treatment of SSc and other fibrotic diseases.
Material and methods
Patients and fibroblast cultures
Fibroblast cultures were obtained from skin biopsies of clinically involved skin from SSc patients (n=9). All patients fulfilled the criteria for SSc as suggested by LeRoy and Megsger.17 The median age of SSc patients was 50 years (range 24–75 years) and their median disease duration was 7 years (range 1.5–12 years). Five patients had limited cutaneous SSc and four had diffuse cutaneous SSc. All patients were positive for antinuclear antibodies; three of the patients were positive for anticentromere antibodies and two were positive for anti-topoisomerase-1 antibodies. None of the patients was treated with immunosuppressive or other potentially disease-modifying drugs at the time of biopsy. Control fibroblasts were obtained from skin biopsies of healthy age and sex-matched volunteers. Fibroblasts were prepared as outgrowth cultures from skin biopsies and cultured as described.18 Fibroblasts from passages 4–8 were used for the experiments. All patients and controls signed a consent form approved by the local institutional review boards.
Selective inhibition of JNK by CC-930
CC-930 was kindly provided by Celgene (Summit, New Jersey, USA). The specificity of CC-930 was proved by extensive biochemical profiling against a panel of 209 protein kinases. At 3 µM, only three human kinases other than JNK showed inhibition that was greater than 80% in the screening panel; MAPK2, epidermal growth factor receptor (EGFR) and mutated EGFR L861Q. Follow-up studies determined IC50 values of 480 nM (MAPK2) and 379 nM (EGFR). The IC50 values of JNK 1, 2, and 3 isomers were 52 nM, 5 nM and 5 nM, respectively.19 Dermal fibroblasts were incubated with CC-930 at concentrations from 0.1 to 10 µM for 24 h. For controls, fibroblasts were incubated with the solvent dimethyl sulphoxide in the same concentrations. In subsets of experiments, fibroblasts were stimulated with recombinant TGFβ at 10 ng/ml or PDGF-BB at 40 ng/ml (both R&D Systems, Abingdon, UK). For stimulation experiments fibroblasts were cultured in Dulbecco's modified essential medium/F12 containing 0.1% fetal bovine serum and ascorbic acid (50 µg/ml) for 3 days before adding cytokines.
MTT assay
The microtitre tetrazolium (MTT) assay was used to assess the metabolic activity of cells incubated with CC-930, as described.20 SSc fibroblasts were incubated with 1 µM CC-930 in 96-well plates for 20 h. Then MTT was added at a final concentration of 1 mg/ml, and the cells were further incubated at 37°C for 4 h. Mock-treated fibroblasts were used as controls, and all other results were normalised to untreated cells.
Western blot analysis
After rinsing twice with phosphate-buffered saline, cells were trypsinised and lysed with Laemmli buffer. Five micrograms of protein from each sample were separated by 10% sodium dodecylsulphate–polyacrylamide gel electrophoresis and electrotransferred onto polyvinylidene difluoride membranes according to standard protocols.21 Immunoblots were incubated with monoclonal antibodies against c-Jun and phospho-c-Jun (Santa Cruz, Heidelberg, Germany). After incubation with horseradish peroxidase-conjugated rabbit anti-mouse antibodies (Dako, Hamburg, Germany), signals were detected with ECL western blotting detection reagents (Amersham Bioscience, Freiburg, Germany). For confirmation of the equal loading of proteins, the amount of β-actin was determined using mouse anti-human β-actin antibodies (Sigma-Aldrich, Munich, Germany).
Immunohistochemistry
Skin biopsies were obtained from clinically involved skin of patients with SSc (n=10). The median age of SSc patients was 45 years (range 21–56 years) and their median disease duration was 4.5 years (range 0.5–6 years). Five patients had limited cutaneous SSc and five patients had diffuse cutaneous SSc. None of the patients was treated with immunosuppressive or other potentially disease-modifying drugs at the time of biopsy. Control biopsies were obtained from age and sex-matched healthy volunteers. Formalin-fixed, paraffin-embedded skin sections were stained with antibodies against phosphorylated c-Jun (Santa Cruz Biotechnology Inc, Heidelberg, Germany). Peroxidase labelled mouse immunoglobulins (Dako, Glostrup, Denmark) were used as secondary antibodies. Irrelevant isotype-matched antibodies were used as controls. Staining was visualised with 3,3-diaminobenzidine tetrahydrochloride (DAB) peroxidase substrate solution (Sigma-Aldrich). Irrelevant isotype-matched antibodies were used as controls. Staining was visualised with DAB peroxidase substrate solution (Sigma-Aldrich).
Quantitative real-time PCR
Total RNA was isolated with the NucleoSpin RNA II extraction system (Machery-Nagel, Düren, Germany) and reverse transcribed into complementary DNA with random hexamers, as described.22 Gene expression was quantified by TaqMan or by SYBR green real-time PCR using the ABI Prism 7700 sequence detection system (PE Applied Biosystems, Rotkreuz, Switzerland) using primers described previously.21 Samples without enzyme in the reverse transcription reaction were used as negative controls to exclude genomic contamination. Unspecific signals caused by primer dimers were excluded by dissociation curve analysis and by no template controls. A predeveloped 18S assay (Applied Biosystems) was used to normalise for the amounts of loaded cDNA. Differences were calculated with the threshold cycle and the comparative threshold cycle method for relative quantification.
Collagen measurements
Total soluble collagen in cell culture supernatants was quantified using the SirCol collagen assay (Biocolor, Belfast, Northern Ireland) as described.23
Bleomycin-induced dermal fibrosis
Dermal fibrosis was induced in 6-week-old DBA/2J mice by injections of bleomycin in defined areas of the upper back.24 One hundred microlitres of bleomycin dissolved in 0.9% sodium chloride (NaCl) at a concentration of 0.5 mg/ml were injected subcutaneously every other day. Subcutaneous injections of 100 μl NaCl were used as controls for treatment with bleomycin. Five different groups of mice with a total number of 36 mice were analysed. One group received injections of NaCl and the other four groups were challenged with bleomycin. Two of the bleomycin-treated groups also received CC-930 in doses of 50 mg/kg and 150 mg/kg by oral gavage twice a day. Another group was also treated intraperitoneally with 50 mg/kg imatinib as a positive control for response to antifibrotic treatment. CC-930 and imatinib were initiated together with the first challenge of bleomycin over 3 weeks. After treatment, animals were killed by cervical dislocation.
TSK 1 mouse model
To investigate the role of JNK in a model of SSc that is less dependent on inflammatory changes than bleomycin-induced dermal fibrosis,10 the antifibrotic potential of CC-930 was evaluated in the tight skin 1 (TSK1) mouse model of SSc. Five groups with a total number of 51 mice were analysed. Four groups consisted of TSK1 mice that were treated with CC-930 at doses of 50 mg/kg or 150 mg/kg, with imatinib at a dose of 50 mg/kg or that did not receive any antifibrotic treatment. The fifth group consisted of control mice of the same genetic background not carrying the TSK1 mutation. The treatment was started at an age of 5 weeks and the outcome was evaluated at an age of 10 weeks.
Mouse model of established dermal fibrosis
To evaluate the regression of fibrosis on inhibition of JNK, a modified model of bleomycin-induced dermal fibrosis was used.25 In this model, treatment was initiated 3 weeks after the beginning of the challenge with bleomycin, when significant dermal fibrosis was already established. The outcome of six different groups with a total number of 40 mice was analysed. The first group of mice received subcutaneous injections of NaCl for 6 weeks. The second group was injected for 3 weeks with bleomycin followed by injections of NaCl for another 3 weeks to analyse the degree of fibrosis before treatment, and to control the spontaneous regression of fibrosis. The third group of mice was killed after 6 weeks of injections with bleomycin. The fourth and the fifth group were treated with CC-930 at doses of 50 mg/kg and 150 mg/kg for the last 3 weeks of continuous challenge with bleomycin for 6 weeks. The sixth group was a positive control group consisting of mice challenged with bleomycin for 6 weeks and treated in parallel with imatinib at doses of 50 mg/kg for the last 3 weeks.
Histological analysis
Skin sections were stained with haematoxylin and eosin for better visualisation of the tissue structure. The dermal thickness was analysed with a Nikon Eclipse 80i microscope (Nikon, Badhoevedorp, The Netherlands) by measuring the distance between the epidermal–dermal junction and the dermal–subcutaneous fat junction at four different skin sections of each mouse.26 The hypodermal thickness was determined by measuring the thickness of the subcutaneous connective tissue beneath the panniculus carnosus at four different sites at the upper back in each mouse.10 The evaluation was performed by two independent examiners.
Detection of myofibroblasts
Myofibroblasts are characterised by the expression of α-smooth muscle actin (αSMA). Fibroblasts positive for αSMA were detected by incubation with monoclonal anti-αSMA antibodies (clone 1A4; Sigma-Aldrich, Steinheim, Germany).27 The expression was visualised with horseradish peroxidase-labelled secondary antibodies and DAB (Sigma-Aldrich). Monoclonal mouse immunoglobulin G antibodies (Calbiochem, San Diego, California, USA) were used for controls.
Hydroxyproline assay
The collagen content of 3 mm punch biopsies from lesional skin samples was determined by hydroxyproline assays as described.28
Statistics
Data are expressed as mean±SEM. The Wilcoxon signed rank tests for related samples and the Mann–Whitney U test were used for statistical analyses. A p value of less than 0.05 was considered statistically significant.
Results
The JNK pathway is activated by pro-fibrotic cytokines and blocked by CC-930
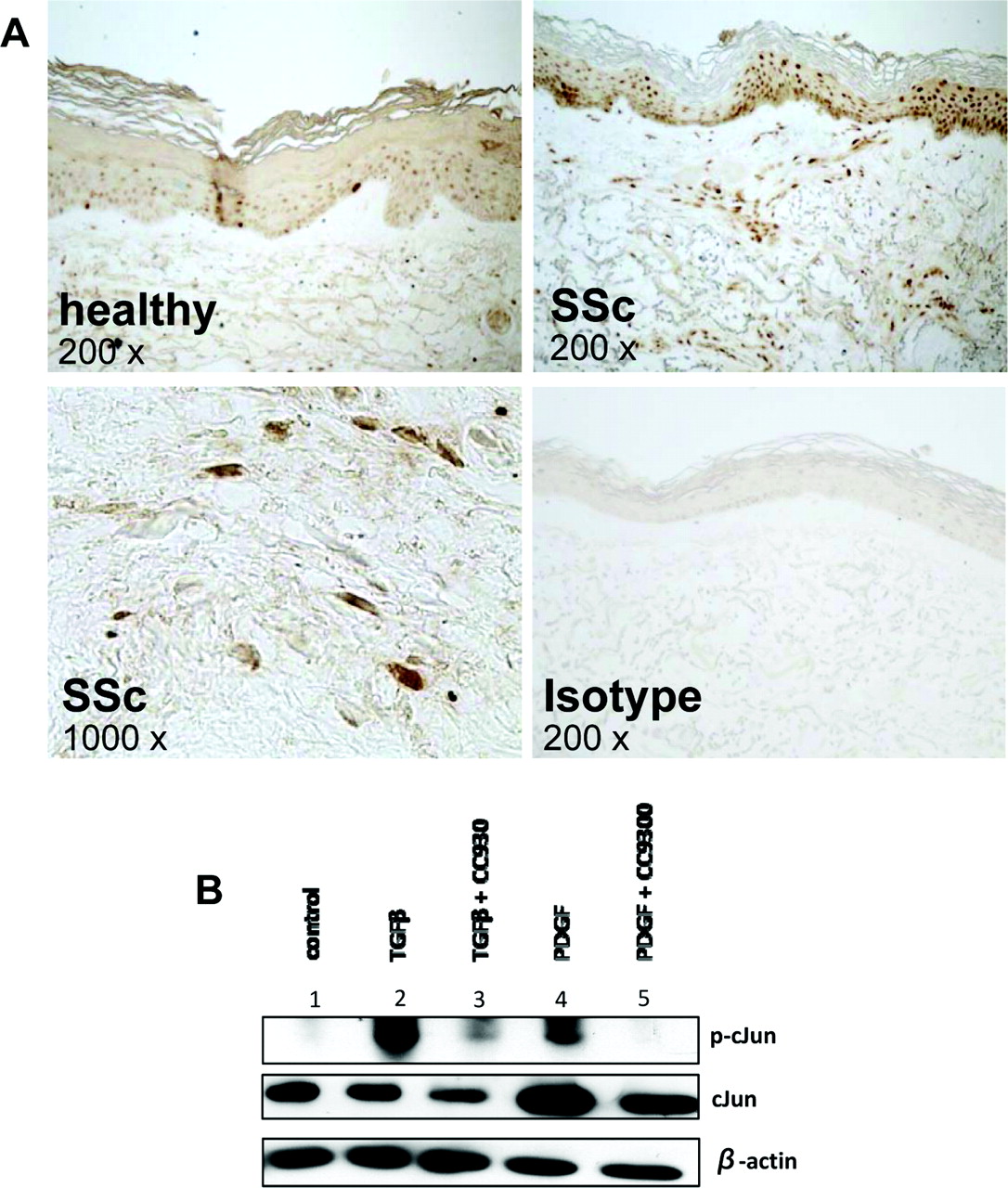
Active JNK phosphorylates the AP-1 transcription factor c-Jun and the phosphorylation of c-Jun is commonly used as a functional read-out for the activity of JNK.29 To assess the activity of JNK in SSc, we performed immunohistochemistry for phosphorylated c-Jun. p-cJun was detected in fibroblasts, vessels and in the epidermis. Increased p-cJun was observed in samples from SSc patients compared with healthy controls. Increased staining was particularly observed in fibroblasts and small vessels of SSc, whereas the differences in the epidermis were less pronounced. (figure 1A). To analyse whether TGFβ and PDGF induce the activation of JNK in SSc fibroblasts, western blot analysis of cultured dermal fibroblasts for c-Jun was performed. Furthermore, we analysed whether the JNK inhibitor CC-930 decreased activation and phosphorylation of c-Jun upon stimulation by TGFβ and PDGF. Stimulation with TGFβ and PDGF potently increased the phosphorylation of c-Jun (figure 1B). In contrast, preincubation with CC-930 at a clinically relevant concentration of 1 μM15 reduced the stimulatory effects of TGFβ and PDGF on the phosphorylation of c-Jun. In particular, CC-930 completely prevented the stimulatory effect of PDGF on c-Jun phosphorylation in fibroblasts (figure 1B).

JNK is activated by TGFβ and PDGF in SSc. (A) Increased levels of phosphorylated c-Jun, the primary substrate of JNK, in SSc patients. Representative images are shown at 200-fold or 1000-fold magnification as indicated. (B) The JNK pathway is activated by pro-fibrotic cytokines, and this activation is blocked by CC-930. To analyse JNK signalling in dermal SSc fibroblasts, the phosphorylation of its target c-Jun was analysed by western blot. Incubation with TGFβ (lane 2) or PDGF (lane 4) increased the protein level of phosphorylated cJun compared with unstimulated fibroblasts (lane 1). Pretreatment of fibroblasts with CC-930 reduced the stimulatory effects of TGFβ (lane 3) and in particular of PDGF (lane 5). Equal loading of proteins was confirmed by quantification of β-actin. JNK, c-Jun N-terminal kinase; p-cJun, phosphorylated cJun; PDGF, platelet-derived growth factor; SSc, systemic sclerosis; TGFβ, transforming growth factor beta.
Inhibition of JNK reduced the synthesis of extracellular matrix in SSc fibroblasts
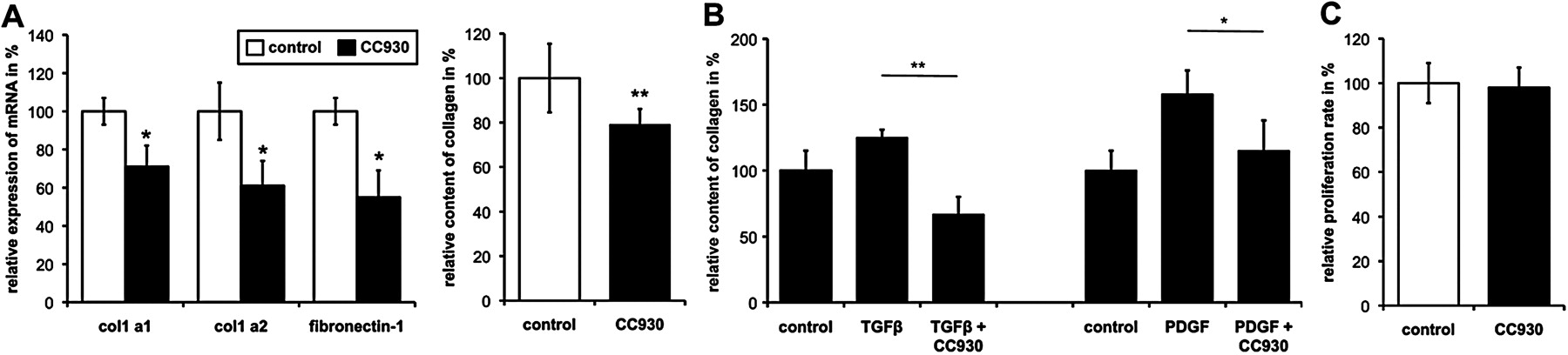
We next investigated whether inhibition of JNK might inhibit the basal release of extracellular matrix proteins from SSc fibroblasts. CC-930 dose-dependently reduced the synthesis of extracellular matrix proteins in SSc fibroblasts. No differences were observed between fibroblasts isolated from patients with limited cutaneous SSc and patients with diffuse cutaneous SSc. Maximal effects were observed at concentrations of 1 µM. At this concentration, CC-930 reduced the messenger RNA levels of col 1a1 and col 1a2 to 71±11% and 61±13%, respectively, and the levels of fibronectin 1 to 55±14% (p<0.02 compared with mock-treated fibroblasts for all) (figure 2A). Consistent with the decreased mRNA levels, incubation with 1 μM CC-930 also reduced the amount of collagen protein in the supernatant to 79±7% (p=0.001) compared with control (figure 2A). In contrast to SSc fibroblasts, no significant effects on the basal collagen synthesis were observed in unstimulated healthy dermal fibroblasts (data not shown).

JNK regulates the release of collagen from fibroblasts. (A) Inhibition of JNK signalling by CC-930 reduced the basal production of extracellular matrix components. Incubation of SSc fibroblasts with CC-930 for 24 h reduced the expression of col 1a1, col 1a2, and fibronectin-1 mRNA and the release of protein into supernatants. The levels of mock-treated fibroblasts were defined as 100%; other results were normalised to this value. Data represent the mean±SEM. *p<0.05; **p<0.01. Stimulation of dermal SSc fibroblasts with TGFβ and PDGF increased the release of collagen into the supernatants. CC-930 completely blocked these stimulatory effects. The collagen levels of untreated fibroblasts were defined as 100%; other results were normalised to this value. Data represent the mean±SEM. *p<0.05; **p<0.01. (B) Inhibition of JNK signalling by CC-930 reduced the stimulatory effects of pro-fibrotic cytokines on collagen production. Stimulation of fibroblasts with TGFβ and PDGF increased the release of collagen into the supernatants. CC-930 completely blocked these stimulatory effects. The collagen levels of untreated fibroblasts were defined as 100%; other results were normalised to this value. Data represent the mean±SEM. *p<0.05; **p<0.01. (C) CC-930 does not exert toxic side effects on dermal fibroblasts. The proliferation rate of cultured dermal SSc fibroblasts was analysed 24 h after incubation with CC-930. No differences could be measured, suggesting CC-930 was not toxic to fibroblasts. The levels of mock-treated fibroblasts were defined as 100%; other results were normalised to this value. JNK, c-Jun N-terminal kinase; PDGF, platelet-derived growth factor; SSc, systemic sclerosis; TGFβ, transforming growth factor beta.
We next analysed whether CC-930 can prevent the pro-fibrotic effects of TGFβ and PDGF in healthy dermal fibroblasts. Incubation with CC-930 at a concentration of 1 μM decreased the stimulatory effects of TGFβ by 230±13% (p=0.005) and of that of PDGF by 73±23% (p=0.04) (figure 2B).
To exclude the possibility that the inhibitory effects on the release of extracellular matrix proteins are due to the toxicity of CC-930 with a general reduction of metabolic activity, MTT assays were performed. CC-930 did not reduce metabolic activity in antifibrotic concentrations, suggesting that inhibition of JNK specifically prevents fibroblast activation and matrix synthesis without toxic side effects (figure 2C).
Inhibition of JNK prevents bleomycin-induced fibrosis
To investigate the role of JNK in inflammation-driven models resembling early inflammatory stages of SSc, the mouse model of bleomycin-induced dermal fibrosis was used. Treatment with CC-930 was well tolerated. No signs of toxicity such as weight loss, reduced activity, or ruffled fur were observed.
Dermal fibrosis with dense accumulation of collagen bundles, inflammatory infiltrates and replacement of the subcutaneous fat tissue by connective tissue was observed in sham-treated bleomycin-challenged mice. In mice treated with CC-930, the development of fibrosis by bleomycin was significantly ameliorated (figure 3A). Treatment with CC-930 reduced dermal thickening in a dose-dependent manner by up to 45±2% at doses of 150 mg/kg (p=0.001). This reduction in dermal thickening was comparable to that observed with imatinib at doses of 50 mg/kg (figure 3A,B). Inhibition of JNK by CC-930 also prevented the accumulation of collagen in lesional skin. The hydroxyproline content of the skin area injected with bleomycin was dose-dependently reduced by up to 53±3% in mice treated with 150 mg/kg CC-930 compared with bleomycin-treated animals (p=0.043) (figure 3C). Consistent with reduced dermal thickening and the decreased accumulation of collagen, inhibition of JNK also inhibited the differentiation of resting fibroblasts into metabolically active myofibroblasts. At doses of 150 mg/kg, the myofibroblast differentiation induced by bleomycin challenge was completely prevented (p=0.001) (figure 3D).

Bleomycin-induced dermal fibrosis was prevented by treatment with CC-930. (A) Dermal fibrosis was strongly reduced by treatment with CC-930. Representative skin sections stained with haematoxylin and eosin are shown at 100-fold magnification. The dermal thickness (B), the hydroxyproline content (C) and the numbers of myofibroblasts (D) were reduced dose-dependently by treatment with CC-930. The efficacy of CC-930 was comparable to that of imatinib, which served as a positive control. The levels of control mice were defined as 100%; other results were normalised to this value. Data represent the mean±SEM. *p<0.05; **p<0.01. NaCl, sodium chloride.
Inhibition of JNK prevents fibrosis in the TSK1 model
To confirm the role of JNK in a less inflammation-dependent model of fibrosis that better resembles the late stages of SSc, we assessed the antifibrotic effects of CC-930 in the TSK1 mouse model. Treatment with CC-930 also exerted potent antifibrotic effects in the TSK1 model, and reduced hypodermal thickening in a dose-dependent manner by up to 85±4% (p<0.001) compared with sham-treated TSK1 mice (figure 4A,B). Consistent with previous reports, imatinib prevented histological changes in TSK1 mice.25 Of interest, the reduction in hypodermal thickening at doses of 150 mg/kg CC-930 was more pronounced than with 50 mg/kg imatinib (figure 4B). Inhibition of JNK by CC-930 also reduced myofibroblast counts in the TSK1 model in a dose-dependent manner. At doses of 150 mg/kg, myofibroblast differentiation was completely prevented, and the myofibroblast counts did not differ from control mice not carrying the TSK1 mutation (p=0.004 compared with untreated TSK1 mice) (figure 4C).

Antifibrotic effects of CC-930 in TSK1 mouse model. (A) Treatment with CC-930 dose-dependently reduced fibrosis in TSK1 mice. Representative skin sections stained with haematoxylin and eosin are shown at 40-fold magnification. Treatment of TSK1 mice with CC-930 reduced hypodermal thickening (B) and completely prevented myofibroblast differentiation (C). The antifibrotic effects of 150 mg/kg CC-930 were comparable with that observed with 50 mg/kg imatinib. The levels of pa/pa mice were defined as 100%; other results were normalised to this value. Data represent the mean±SEM. **p<0.01. TSK, tight skin 1.
Inhibition of JNK can induce regression of established fibrosis
To evaluate whether inhibition of JNK might also be effective for the treatment of established fibrosis, a modified model of bleomycin-induced dermal fibrosis was used. Prolonged injections of bleomycin for 6 weeks resulted in progressive fibrosis with increased dermal thickening to 226±6% compared with a dermal thickening of 175±8% after 3 weeks of bleomycin challenge (p=0.021 compared with NaCl injections for 6 weeks). The dermal thickness in mice treated with CC-930 for the last 3 weeks was significantly reduced to 127±4% at doses of 150 mg/kg (p=0.008 compared with bleomycin for 6 weeks) (figure 5A,B). CC-930 thus decreased the level of dermal thickness under the pretreatment level (127±4% vs 175±8% compared with NaCl injections for 6 weeks, p=0.008 and p=0.021). Besides dermal thickness, the number of myofibroblasts was strongly reduced on treatment with 150 mg/kg CC-930 under the pretreatment level (171±31% vs 443±15% compared with NaCl injections for 6 weeks, p=0.012 and p=0.014) (figure 5C). In addition, the accumulation of collagen assessed by hydroxyproline assay was significantly reduced in mice treated with 150 mg/kg CC-930 for the last 3 weeks of continuous bleomycin challenge for 6 weeks (p=0.025) (Figure 5D). The inhibition of JNK thus not only prevents the development of fibrosis in different models, but can also induce the regression of pre-existing fibrosis.

CC-930 induced regression of experimental fibrosis. (A) Treatment with CC-930 did not only stop further progression, but also induced regression of bleomycin-induced dermal fibrosis. Representative skin sections stained with haematoxylin and eosin are shown at 100-fold magnification. Treatment with CC-930 reduced dermal thickening (B), hydroxyproline content (C) and myofibroblast counts (D) to below pretreatment levels despite ongoing challenge with bleomycin. The level of mice receiving sodium chloride injections for 6 weeks was defined as 100%; other results were normalised to this value. Data represent the mean±SEM. *p<0.05; **p<0.01. NaCl, sodium chloride.
Discussion
In this paper we highlight an important role of JNK in experimental fibrosis and demonstrate that JNK signalling is activated by pro-fibrotic cytokines. TGFβ and PDGF, two key players in fibrotic disease activate JNK in SSc fibroblasts, as demonstrated by phosporylation of its downstream target c-Jun in vitro and in vivo. Consistent with our results in fibroblasts, an increased phosphorylation of JNK on stimulation with TGFβ has also been demonstrated in tracheal epithelial cells and in breast cancer cells.8 9 These findings demonstrate that JNK is a common target of pro-fibrotic mediators and indicate a role of JNK as intracellular mediators of the pro-fibrotic effects of TGFβ and PDGF.
Indeed, selective inhibition of JNK by CC-930 potently reduced the activation of fibroblasts. CC-930 inhibited the release of extracellular matrix proteins. Inhibition of JNK also prevented experimental fibrosis in the bleomycin model of early inflammatory stages of SSc. Moreover, inhibition of JNK also induced the regression of pre-established experimental fibrosis. The mouse model of bleomycin-induced fibrosis is highly dependent on inflammation and this results in hypersensitivity to anti-inflammatory therapies. CC-930 also exerted potent antifibrotic effects in the TSK1 model of later, non-inflammatory, stages of SSc. In contrast to bleomycin-induced fibrosis, inflammatory infiltrates are not observed in the TSK1 model and TSK1 mice are not considered to be hyperresponsive to anti-inflammatory therapies in general. However, B cells seem to play a very central role in the development of fibrosis in TSK1 mice, and the TSK1 model may overestimate the effects of B-cell targeting therapies. These limitations should be considered when interpreting the results obtained in individual models. However, a prudent combination of different models may overcome many of the limitations of individual animal models. In the case of targeting JNK, additional evidence of antifibrotic effects is provided from models of other fibrotic disorders. Consistent with our data obtained with CC-930, JNK-/- mice are protected from liver inflammation and subsequent fibrosis induced by a choline-deficient L-amino acid-defined diet.30 Deficiency of JNK also prevented subepithelial collagen deposition in an ovalbumin-driven model of allergic airways disease.31 Together, these findings suggest that JNK plays crucial roles in the pathogenesis of fibrotic diseases.
Several potential targets for translational therapies of SSc have recently been identified (figure 6). Some of them target similar pathways at different levels. Tyrosine kinase inhibitors such as imatinib, inhibitors of Src kinases, Jak-2 inhibitors and also JNK inhibitors are all growth factor signalling at different levels and may exert their antifibrotic effects by the inhibition of intracellular cascades that are activated by TGFβ and PDGF.20 24 25 32 33 The antifibrotic effects of peroxisome proliferator-activated receptor gamma agonists may also primarily be mediated by the inhibition of TGFβ signalling.34 35 Binding of serotonin to 5-HT2B receptors stimulates the release of TGFβ. The inhibition of 5-HT2B may thus exert its antifibrotic effects indirectly by decreased TGFβ signalling.36 The morphogenic pathways such as Wnt, Hedgehog and Notch, which have recently been implicated in fibrosis, interact with each other and with TGFβ signalling during development.7 29 37,–,40 Although direct links between these pathways and TGFβ signalling in fibrosis have not yet been established, altered TGFβ signalling may contribute to the pro-fibrotic effects of Wnt, Hedgehog and Notch pathways. Antibodies against interleukin (IL)-4 and IL-13 inhibit other pro-fibrotic cytokines, but also directly target fibroblast activation.41 In contrast, IL6 receptor antagonists, cannabinoid receptor 1 antagonists and cannabinoid receptor 2 agonists primarily target fibroblast activation indirectly by inhibiting the release of pro-fibrotic mediators from inflammatory cells.26 27

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Proposed mechanism of action of novel antifibrotic therapies and potential interactions. JNK, c-Jun N-terminal kinase; NCID, Notch intracellular domain; PDGFR, platelet-derived growth factor receptor; PPARγ, peroxisome proliferator-activated receptor gamma; RTK, receptor tyrosine kinase; TCF, ; TGFR, transforming growth factor β receptor.
We did not observe toxic effects of CC-930 in antifibrotic concentrations. Inhibition of JNK did not reduce the metabolic activity of cultured fibroblasts. Mice treated with CC-930 did not show any signs of toxicity such as weight loss, reduced activity, or ruffled fur. In an extension of our preclinical data, phase III clinical trials with different inhibitors of JNK for the treatment of rheumatoid arthritis and different types of cancer showed that inhibition of JNK is well tolerated in humans.16 The first data on CC-930 in humans are also available. Results from dosing studies in healthy volunteers have indicated that CC-930 is well tolerated and exposure is dose-proportional.15 Moreover, no unexpected drug-dependent effects were observed from phase I clinical assessment.15 These data suggest that inhibition of JNK by inhibitors such as CC-930 is well tolerated, and that a potential application in patients with fibrotic diseases might not be hindered by toxicity.
In summary, we demonstrate that JNK is activated by pro-fibrotic cytokines and regulates the synthesis of extracellular matrix proteins. Selective inhibition of JNK by CC-930 prevented skin fibrosis in different models resembling different stages of SSc, and induced regression of pre-established experimental fibrosis. As inhibition of JNK is well tolerated and inhibitors are currently being evaluated in clinical trials for other indications, JNK might be an interesting molecular target in SSc and other fibrotic diseases.
Acknowledgments
The authors would like to thank Maria Halter and Anna-Maria Herrmann for excellent technical assistance.
References
Footnotes
-
Funding This study was funded by grants DI 1537/1-1 and DI 1537/2-1 of the Deutsche Forschungsgesellschaft, the Career Support Award of Medicine of the Ernst Jung Foundation, grants A20 and A40 of the Interdisciplinary Center of Clinical Research in Erlangen, a grant from Celgene and the CMH research project no 00000023728.
-
Ethics approval Ethics approval was obtained from the local institutional review boards.
-
Patient consent Obtained.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.