Article Text

Abstract
Background Cryopyrin-associated periodic syndromes (CAPS) consist of a continuum of autoinflammatory diseases caused by a defect in interleukin 1β regulation. Although symptoms may vary widely, the discovery, in 2001, of the gene involved (NLRP3) has dramatically helped diagnosis.
Objectives To define the spectrum and prevalence of NLRP3 mutations in France and to delineate initial criteria before molecular analysis.
Methods Retrospective review (2001–9) of genetic analysis data and request forms of patients living in France with an NLRP3 mutation since the set up of CAPS molecular diagnosis by the three French laboratories providing this test (GenMAI network).
Results Over 800 analyses of this gene have been conducted, identifying 135 cases with an NLRP3 mutation (55 probands; 33 multiplex families); the estimated prevalence in France was equal to 1/360 000. A total of 21 different sequence variants were detected, among which four are common and nine are new mutations.
Conclusions Although the number of NLRP3 test requests has doubled over the past 5 years, genetic screening has not contributed to enhanced detection of new index cases each year. There are two possible reasons for this: (i) no clinical prerequisite for genetic diagnosis and (ii) few new large families are now identified (unlike the initial study based on a selection by linkage). A set of initial clinical criteria have been drawn up which it is recommended should be fulfilled before a patient is tested: at least three recurrent bouts, age at disease onset < 20 years and elevated levels of C-reactive protein, especially in individuals with urticaria and moderate fever.
Statistics from Altmetric.com
Introduction
Cryopyrin-associated periodic syndromes (CAPS) consist of rare, autoinflammatory disorders,1 which are a continuously expanding group of conditions caused by defects in genes acting in or regulating innate immunity signalling pathways.2 CAPS are usually dominantly inherited. The CAPS gene, NLRP3 (nucleotide-binding domain, leucine-rich repeat family, pyrin domain containing 3), previously named CIAS1 (cold-induced autoinflammatory syndrome 1) was identified in 20013 following genetic linkage to 1q44 in multiplex families.4 5 Sporadic cases due to de novo mutations have also been identified. The CAPS protein, cryopyrin, is a key inflammasome component.6 The inflammasome is a multimeric protein complex that triggers activation of the caspase 1 enzyme, which in turn catalyses the cleavage of pro-interleukin 1β (IL-1β) into its active form IL-1β, a very potent proinflammatory and pyrogenic cytokine. The online database dedicated to hereditary autoinflammatory diseases (Infevers) has recorded 118 sequence variants in NLRP3 so far that are all point mutations.7 All but three reported CAPS mutations are located in exon 3 that encodes the oligomerisation (NLR binding) domain of cryopyrin. The three other mutations are located in exons 4 and 6, in the leucine-rich repeat domain.8,–,12 NLRP3 missense mutations are probably gain of function mutations that cause constitutive activation of IL-1β signalling.
The discovery of this gene linked three different phenotypes that were initially described independently.3 13 Familial cold autoinflammatory syndrome (FCAS) is the mildest form, mainly presenting as cold-induced recurrent bouts of fever associated with urticaria and arthralgia. In Muckle–Wells syndrome (MWS), these symptoms are often complicated with neurosensorial deafness and renal AA amyloidosis. Chronic infantile neurological cutaneous and articular syndrome (CINCA), also called NOMID (neonatal onset multisystem inflammatory disease), is the most severe form of CAPS, as it includes all the symptoms observed in MWS, its onset is neonatal and the clinical picture may include bone deformation and dysmorphy, chronic meningitis, blindness and mental retardation. This classification is helpful for clinicians, but it becomes clear, through the identification of patients with overlapping symptoms, that CAPS should be considered as a clinical continuum rather than as a group of three delimited phenotypes.8
Therefore, because CAPS include such a broad spectrum of clinical presentations, no clinical criteria can be used for a general definition of these syndromes. At most, some signs, such as urticaria that are more common in CAPS than in other hereditary recurrent fevers are often regarded as indispensable to the diagnosis. For these reasons, the current denomination of CAPS reflects exclusively the pathophysiological basis of this group of conditions that includes all the disease phenotypes associated with mutations in the NLRP3 gene. However, it should be noted that while some mutations are clearly involved in the disease, others (eg, p.Val198Met), remain controversial, which makes it even more difficult to assess the impact of this gene in the symptomatology presented by the patients. Nevertheless, confirmation of the involvement of this key protein of the inflammasome in the patients' phenotype is of great importance given the therapeutic efficacy of anti-IL-1 drugs.14 15
In 2004, the French Ministry of Health decided to support patients with an orphan disease and health professionals involved in rare diseases by creating clinical patient care designated centres and networks for genetic diagnosis. Clinical centres are organised into reference (expertise) and competence (proximity care) centres. Our three university hospital laboratories created the French network for the genetic diagnosis of autoinflammatory diseases (named GenMAI, http://genmai.chu-montpellier.fr/EN/index.html) that performs all CAPS genetic testing in France (as recorded by the European database for rare diseases, http://www.orpha.net/consor/cgi-bin/index.php?lng=EN). We work closely with the corresponding designated clinical centres.
Patients with CAPS have been occasionally identified in numerous countries.4 8 16,–,21 Here, we describe the epidemiological and genetic characteristics of all French patients bearing a mutation in the CAPS gene (N=135). We also tried to retrospectively delineate the clinical picture in positive patients that led doctors to order this molecular analysis, with a special focus on urticaria, which is often considered a key symptom in CAPS.22 Since CAPS are a clinical continuum and this study was conducted within a genetic network, our aim was to evaluate and improve the indication for genetic testing, rather than identify phenotype genotype correlation. This series is the largest reported so far.
Patients and methods
Study design and data protection
This study presents the merged data of the GenMAI network since the molecular test was developed. We retrospectively reviewed the genetic analysis request forms of all patients who were discovered to carry an NLRP3 mutation. This form contains information about the referring clinicians, the patient's epidemiological data and a list of clinical and biological symptoms frequently encountered in autoinflammatory disorders. A survey focusing on cutaneous signs was also undertaken.
All patient identity data was numerically encoded by each centre, homogenised and entered anonymously into a password-protected database (Microsoft Access) created specifically by the coordinator of the study (IT) for this purpose.
Genetic analysis
Genetic testing of all French patients suspected of having CAPS was performed in one of the three GenMAI laboratories. All patients or their guardians gave signed informed consent. The first step of the mutation screening strategy consisted of sequencing the NLRP3 exon 3 (forward and reverse strands) in all three laboratories, since this region harbours 97% of the known CAPS mutations.7 Exon 3 was screened for 821 patients; the other exons and intronic boundaries were analysed in a subset of patients following a specific request of the prescribing doctor in patients who they considered particularly clinically convincing (N=52).
Results
Performance and distribution of the genetic analysis requests
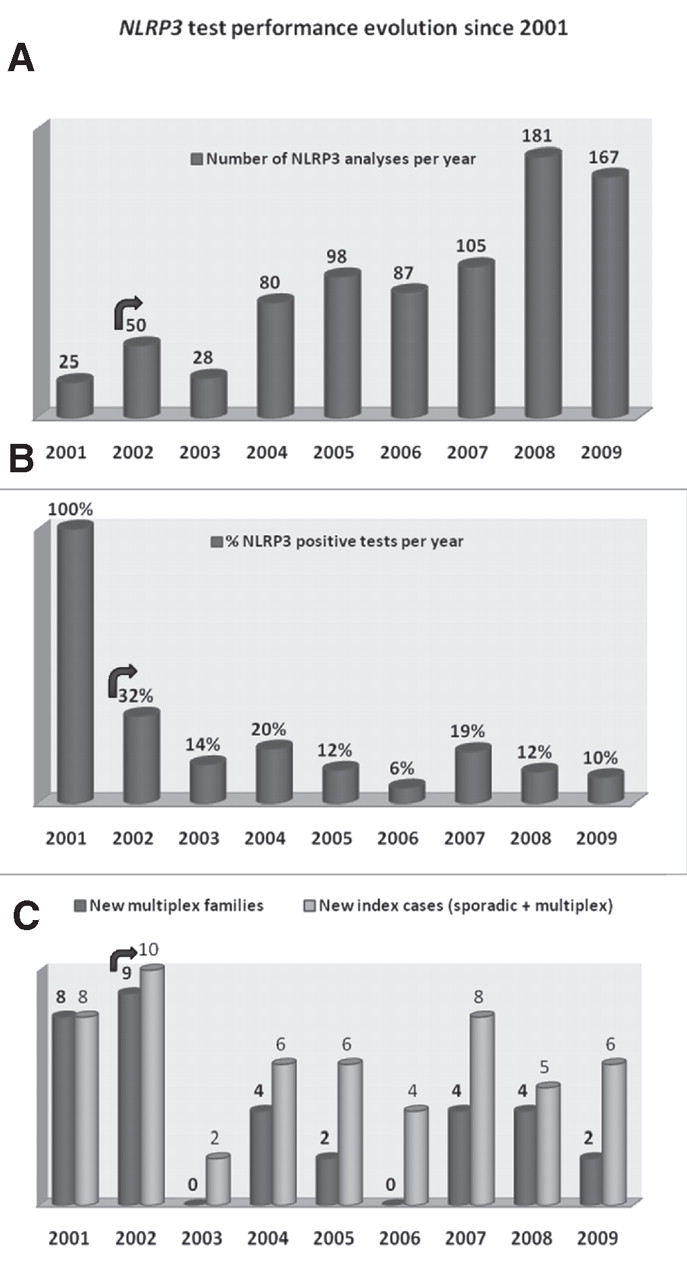
We identified one NLRP3 mutation in 135/821 individuals investigated (16%). A study of the number of test requests since 2001 shows that it has doubled over the past 5 years (figure 1A). The percentage of positive cases per year dropped rapidly after the discovery of the gene, ranging from 6% to 32% (figure 1B).

NLRP3 test performance evolution since 2001. The total number of French patients with mutations was assessed from merged data from the GenMAI network. (A) Total number of NLRP3 tests per year since 2001. Note that it doubled since 2004. (B) Performance of the tests. This positive rate has been quite stable for 1 year after the gene discovery. (C) Comparison of the total number of positive index cases (sporadic and multiplex families) with the number of positive new multiplex families. The arrow shows the start of genetic diagnosis in the network after discovery of the gene.
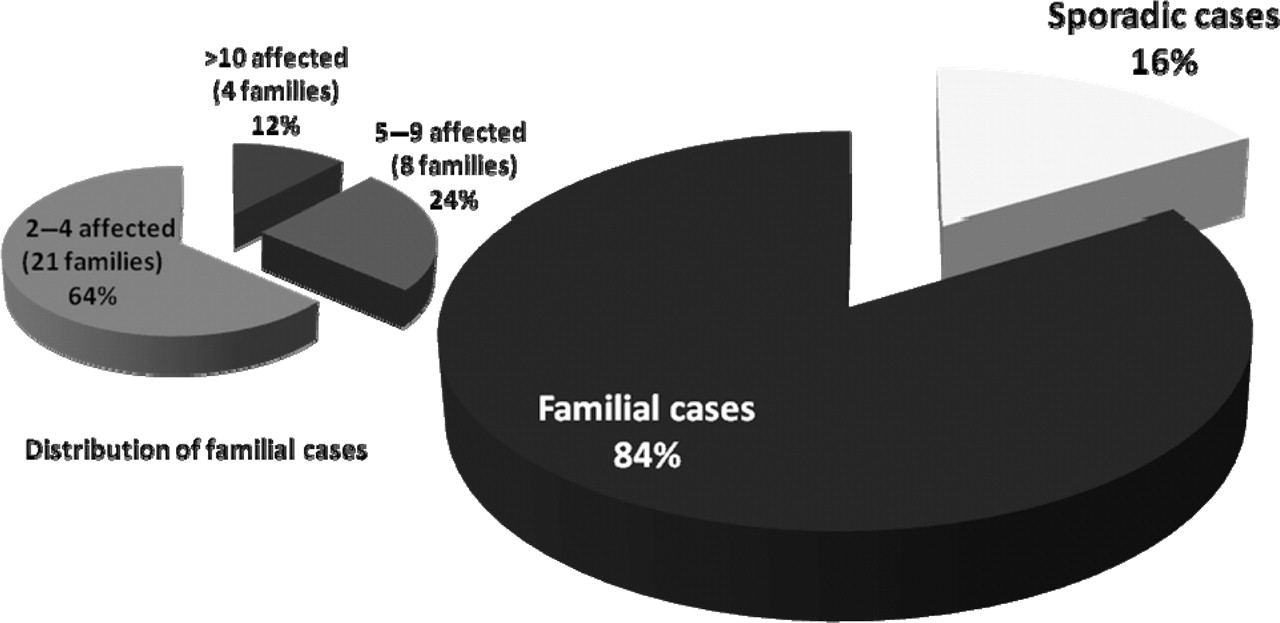
NLRP3 mutations were found in 55 unrelated index cases. Examination of the familial distribution of the 135 patients with the NLRP3 mutation showed that 113 (84%) belonged to 33 multiplex families, and 22 (16%) were referenced as sporadic cases (figure 2). Four families were particularly large (10 or more cases). Since 2003, only 2–8 new index cases have been identified each year in France, corresponding to 0–4 new multiplex families and 1–4 sporadic cases (figure 1C).

Familial structure of the NLRP3-positive cases in France. The 135 positive cases included 22 sporadic (16%) and 113 familial cases (84% belonging to 33 families). Twelve families had more than eight affected individuals. Seven of the 22 sporadic cases had the debated p.Val198Met mutation.
Demographic data
The patients recruited resided across France (online supplementary figure 1). Forty-four per cent of the patients were referred by either reference or competence clinical centres dealing with autoinflammatory diseases. The referring doctors were mainly from internal medicine (40%), rheumatology (17%) or paediatric (16%) centres.
We explored the demographic data of these genetically positive patients. The male to female ratio was 0.88, supporting the absence of a gender effect on the disease. When the patient's origin was indicated (half of the charts), we noticed that all were of Caucasian origin.
From the study of genealogical trees in the identified families we estimated the total number of individuals carrying a NLRP3 mutation at around 180, which includes patients who were genetically confirmed, and the reported related symptomatic cases who were not available for genetic testing. This allowed us to calculate the minimal prevalence of NLRP3 in France as 1/360 000 (current number of inhabitants in France 64.3 millions).
Distribution of the NLRP3 mutations
We found 21 different sequence variants, all being missense mutations (figure 3). Mutation p.Thr348Met was the most common, accounting for 11 (20%) of the 55 families. The second most common mutation was the debated p.Val198Met (seven sporadic cases and two small multiplex families in which the segregation was consistent with the phenotype). In one family, the father and the son presented CAPS symptoms, but p.Val198Met was only found in the father (not shown). The third most common mutation, p.Arg260Trp, was found in eight families (15%), and the fourth one, p.Ala439Val, in five families (9%). A newly identified mutation p.Glu311Lys was found in two unrelated families (4%), one simplex and one with two affected sisters.

Distribution of the NLRP3 mutations in France. The pie chart individualises the seven most common mutations. Other—14 mutations were found in only one family. Six have already been reported in non-French patients (p.Asp303His, p.Ala352Val, p.Leu353Pro, p.Thr436Asn, p.Glu525Lys, p.Gly569Arg), but the eight others (p.Arg168Gln, p.Val262Gly, p.Gly301Asp, p.Ile313Val, p.Ile334Val, p.Ala439Thr, p.Glu690Lys, p.Met701Thr) are new. The asterisk indicates that the p.Val198Met is a debated variant.
Fourteen mutations were found in only one family. Six have been already reported in other countries (p.Asp303His, p.Ala352Val, p.Leu353Pro, p.Thr436Asn, p.Glu525Lys, p.Gly569Arg). The other eight were new and segregated in multiplex (3–12 cases) families (p.Val262Gly, p.Gly301Asp, p.Ile313Val, p.Ala439Thr), or were found in small families (<3 cases) or sporadic cases (p.Arg168Gln, p.Ile334Val, p.Glu690Lys, p.Met701Thr). Only one non-exon 3 mutation, p.Tyr859Cys in exon 6, was detected in two unrelated multiplex families.
DNA from the two healthy parents was available in three of the 22 sporadic cases. We found that mutations p.Asp303His, p.Glu311Lys and p.Thr348Met occurred de novo in probands (not shown).
Calling symptoms in NLRP3-positive patients
The symptoms recorded in the forms were compatible with FCAS (39% of the patients), MWS (63%), CINCA (19%), or were not documented (10%). The most common symptoms were elevated C-reactive protein (CRP) values, articular, ocular and cutaneous manifestations, each present in over 85% of the documented cases (figure 4A). The age at onset was recorded in 113/135 cases and was found to be ≤10 years in 86% of the cases, and ≤20 years in 96% (Figure 4B). The 4% of remaining cases (N=5) in whom the disease manifested after the age of 20 carried the debated p.Val198Met mutation (N=3), or one of two novel mutations (p.Ile313Val and p.Met701Thr). Where noted, the triggering factor was change of temperature in 98% of the cases, mainly coldness (not shown). Fever was absent in 34% of the patients, and not mentioned or not quantified in another 40% suggesting that it was probably not highly elevated in the latter two groups (figure 4C). When quantified, fever reached or exceeded 39°C in only 13% of all the patients.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Clinical features of the patients with an NLRP3 mutation in France. (A) Phenotype features described in the genetic analysis request form. Cutaneous signs (urticaria, oedema, folliculitis, maculopapula) were recorded after a second specific survey. Articular signs included arthritis and arthralgia; ocular signs included uveitis, papillitis and conjunctivitis; abdominal signs included pain, vomiting and diarrhoea; triggering factors included cold, heat, fatigue and stress, thoracic signs included pain, pericarditis and pleuritis. (B) Distribution of age at disease onset. The 4% of patients with age at onset >20 had either novel (p.Ile313Val, p.Met701Thr) or p.Val198Met mutations. (C) Distribution of the temperature presented by the patients during attacks.
Since urticaria is widely thought to be quite specific to CAPS among hereditary recurrent fevers, we exhaustively sought information on cutaneous symptoms. We thus contacted all referring clinicians who did not originally document this item in the test request form to ascertain the absence of urticaria, or update our data if necessary. We found that 87% (117/135) of the patients manifested this type of cutaneous rash, with or without additional skin manifestations such as oedema, folliculitis, papulo-macula, erythema nodosa or ichthyosis. All patients with the most frequent and unambiguous mutations (eg, p.Arg260Trp, p.Thr348Met, p.Ala439Val) had urticaria, whereas this symptom was not constant in several of the patients with debated (p.Val198Met) or rare (p.Glu311Lys, p.Glu690Lys, p.Met701Thr, p.Tyr859Cys) mutations.
Discussion
This study describes the constellation of NLRP3 mutations in all French patients referred for suspicion of CAPS. We identified 135 positive individuals, which represents the largest series reported so far. The information retrieved on affected individuals allowed us to estimate for the first time the prevalence of NLRP3-associated diseases in France as 1/360 000 people.
We detected a total of 21 different mutations, all simple base substitutions. Four of them (p.Thr348Met, p.Arg260Trp, p.Ala439Val, p.Asp303Asn) are definitely causative mutations since they have been repeatedly reported and always segregated with the disease in this study. These four mutations account for 51% of this French series, and are also four of the most common mutations in other populations, with a similar phenotypic distribution.8 Another frequently seen sequence variant that we found in nine index cases is p.Val198Met. Our data tend to corroborate the conclusion that p.Val198Met alone is not responsible for CAPS, but rather a non-deleterious polymorphism, or a modifier. Indeed, its allele frequency was found to be 0.0055 (9/1642) in our patients and 0.0074 in asymptomatic Caucasians.8 In one previously reported family,23 the individuals were symptomatic only if they inherited a second mutation of unclear significance in another hereditary recurrent fever gene—that is, p.Arg121Gln (known as R92Q) in the TNFRSF1A gene, suggesting digenic transmission for these debated mutations. In another family reported herein, mutation p.Val198Met was only found in the father, whereas both the father and his son were symptomatic, supporting the hypothesis that another cause is responsible for the disease.
Nine of the 21 mutations were new (figure 3). Mutation p.Glu311Lys was found in two small unrelated families and p.Val262Gly, p.Gly301Asp, p.Ile313Val and p.Ala439Thr segregated with the phenotype, arguing for a causative role of these five mutations in the disease. Additional reports, development of biochemical tests and appropriate in vitro functional assays would also help to confirm pathogenicity of p.Arg168Gln, p.Ile334Val, p.Glu690Lys and p.Met701Thr as they were found in single sporadic cases or in small families.
We identified 22 (16%) apparently sporadic cases, and could confirm de novo mutations in three of them, underlining that it may be worth testing the parents. Seven of the 22 sporadic patients had p.Val198Met (5%). Considering that this variant is probably not a causative mutation, the rate of 16% sporadic CAPS cases is probably overestimated in France.
Genetic tests are increasingly ordered as information about the pathophysiology of rare diseases is becoming widely available. One of the tasks of French networks for the genetic diagnosis of rare diseases is to improve test quality and performance. In this study, we highlighted the fact that no mutation was found in 84% of the overall genetic diagnosis tests requested. In 2001, the rate of positive patients reached 100% (figure 1B) because these French families corresponded to those selected through genetic linkage during the initial genome round for the CAPS gene localisation.4 18 This test performance is now much lower and quite stable (mean 12% since 2003). The number of tests ordered doubled from 2003, reaching 167 in 2009, but this was associated with an increase in the number of diagnosed cases per family and not with an increase in the number of new index cases identified per year (average N=6, multiplex plus sporadic). Altogether, these data show that extending indication of NLRP3 screening did not contribute to genetic confirmation of new index cases.
These observations prompted us to draft the initial constellation of symptoms that led French clinicians to recommend NLRP3 gene analysis in patients in whom we found a mutation. Gattorno et al24 recently provided a clinical score before genetic analysis for three of the hereditary recurrent fevers that did not include CAPS. We suggest that a genetic analysis of the NLRP3 gene should only be ordered as a first-line test for patients who comply with the following minimal prerequisites (especially if they represent sporadic cases)—that is, at least three recurrent bouts to limit common causes such as infections, age at disease onset <20 years and elevated levels of CRP as they were prevalent in 96% and 91% of the cases, respectively. Indeed, when combined, early age at onset and biological inflammation were present in 89% of the patients. However, since CRP levels were not frequently documented, the predictive value of these combined items should be confirmed in a prospective study.
As expected from clinical descriptions of CAPS, urticaria was present in the majority of patients (87%) and reached 100% in patients with the four most common and unambiguous mutations. Ocular signs and temperature variation as a triggering factor may also represent relevant inclusion clinical signs. Fever, a main symptom basically found in the other recurrent fevers, was however found to be relatively moderate in CAPS, a feature that has not been emphasised before. Our study also shows that atypical cryopyrinopathies might be associated with rare NLRP3 sequence variants (eg, urticaria almost absent in patients with p.Tyr859Cys). Conversely, patients with a phenotype compatible with CAPS from the clinical charts remained a mutation orphan. These conditions might be caused by NLRP3 mutations in other exons and we recommend that this screening should be performed in all exon 3-mutation-negative patients with convincing clinical history. Alternatively, molecular mechanisms other than mutations in NLRP3 coding regions may be involved, such as in introns or the promoter region, intragenic or chromosomal genetic rearrangements or mutations in other genes (eg, NLRP12). However, the search for such new molecular mechanisms is difficult in routine settings.
In conclusion, the epidemio-genetic study of patients carrying an NLRP3 mutation conducted here showed that new mutations are still detectable, but a better indication of test requests is warranted. We suggest that genetic diagnosis should be first aimed at patients with recurrent bouts of biologically proven inflammation, occurring before the age of 20, especially in individuals with urticaria and moderate fever.
Acknowledgments
The authors would like to thank Britt House for editing the English.
References
Supplementary materials
Web only data
Files in this Data Supplement:
Footnotes
-
The French CAPS study group B Bader-Meunier, JM Berthelot, F Cartault, F Caux, J Chevrant-Breton, MP Cordier, R Dhote, A Duquesne, C Frances, P Frange, D Gaillard, B Granel, G Grateau, E Hachulla, PY Hatron, V Hentgen, C Jorgensen, N Kacet, G Kaplanski, I Koné-Paut, D Lacombe, C Lejeune, I Lemelle, S Marlin, A Meyrier, S Morell-Dubois, B Neven, J Ninet, JB Philit, P Quartier, J Stirnemann, R Touraine.
-
Funding This work was supported by the French Ministry of Health, the Centre Hospitalo-Universitaire de Montpellier, the Assistance Publique des Hôpitaux de Paris and Novartis Pharma SAS.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Miscellaneous