Article Text

Abstract
Background Rheumatoid arthritis (RA) can be divided into two major subsets based on the presence or absence of antibodies to citrullinated peptide antigens (ACPA). Until now, data from genome-wide association studies (GWAS) have only been published from ACPA-positive subsets of RA or from studies that have not separated the two subsets. The aim of the current study is to provide and compare GWAS data for both subsets.
Methods and results GWAS using the Illumina 300K chip was performed for 774 ACPA-negative patients with RA, 1147 ACPA-positive patients with RA and 1079 controls from the Swedish population-based case–control study EIRA. Imputation was performed which allowed comparisons using 1 723 056 single nucleotide polymorphisms (SNPs). No SNP achieved genome-wide significance (2.9 × 10–8) in the comparison between ACPA-negative RA and controls. A case–case association study was then performed between ACPA-negative and ACPA-positive RA groups. The major difference in this analysis was in the HLA region where 768 HLA SNPs passed the threshold for genome-wide significance whereas additional contrasting SNPs did not reach genome-wide significance. However, one SNP close to the RPS12P4 locus in chromosome 2 reached a p value of 2 × 106 and this locus can thus be considered as a tentative candidate locus for ACPA-negative RA.
Conclusions ACPA-positive and ACPA-negative RA display significant risk allele frequency differences which are mainly confined to the HLA region. The data provide further support for distinct genetic aetiologies of RA subsets and emphasise the need to consider them separately in genetic as well as functional studies of this disease.
This paper is freely available online under the BMJ Journals unlocked scheme, see http://ard.bmj.com/info/unlocked.dtl
Statistics from Altmetric.com
Introduction
Rheumatoid arthritis (RA) is a common inflammatory joint disease caused by a complex interplay of genetic variants and environmental exposures.1,–,3 Disease outcomes in RA are highly variable, and the presence or absence of antibodies to citrullinated peptide antigens (ACPA) has proved to be one of the best clinical predictors of the severity of disease course.4 5 In addition, ACPA-positive patients with unspecified arthritis respond differently from ACPA-negative patients with RA to early methotrexate therapy.6
In recent years a number of candidate genes have been shown to associate differently with ACPA-positive and ACPA-negative RA. Several genetic variants within the HLA region, specifically the shared epitope-containing HLA-DRB1 alleles,7,–,9 PTPN22 alleles,10 11 as well as a variant in the C5-TRAF1 region,12 13 TNFAIP3,14 15 CD40, CCL21 and many other loci16 17 have been shown to associate with ACPA-positive RA but have not been tested for ACPA-negative RA. By contrast, variations in IRF518 and C-type lectin genes19 appear to be associated with ACPA-negative RA. Association of ACPA-negative disease with HLA-DRB1*03 haplotype was previously suggested20 21 but has not been replicated in a larger study.22 In at least one study23 an indication for an association of PTPN22 marker with ACPA-negative RA was presented based on 65 cases of RA. On the other hand, STAT4 variant has been shown in a meta-analysis to be a risk factor for both subgroups of RA.24 Smoking is the only environmental risk factor unambiguously associated with the risk of RA, but it too appears to affect risk only for ACPA-positive patients.8
These data on different risk factors for ACPA-positive and ACPA-negative RA have been used to propose a new aetiological model for ACPA-positive RA, whereas no such model yet exists for ACPA-negative disease.18 25 26 A major implication of these observations is that genetic and immunological studies of RA should consider this heterogeneity of RA.
So far, the most powerful technique to analyse the effects of genetic variation on disease susceptibility—that is, the genome-wide association study (GWAS)—has not addressed this heterogeneity and genome-wide data published on RA to date have either considered ACPA-positive disease alone13 17 27 or grouped both subtypes together.14 28
In order to provide a more complete picture of genetic risk factors for RA, we have performed genome-wide association analyses in both RA subsets in two different collections of RA cases and controls defined by ACPA status (Swedish Epidemiological Investigation of Rheumatoid Arthritis (EIRA) and North American RA Consortium (NARAC)), and in data from the Wellcome Trust Case–Control Consortium (WTCCC) which contains patients from both subsets but where subdivision according to ACPA status has not yet been performed.
Methods
Subjects
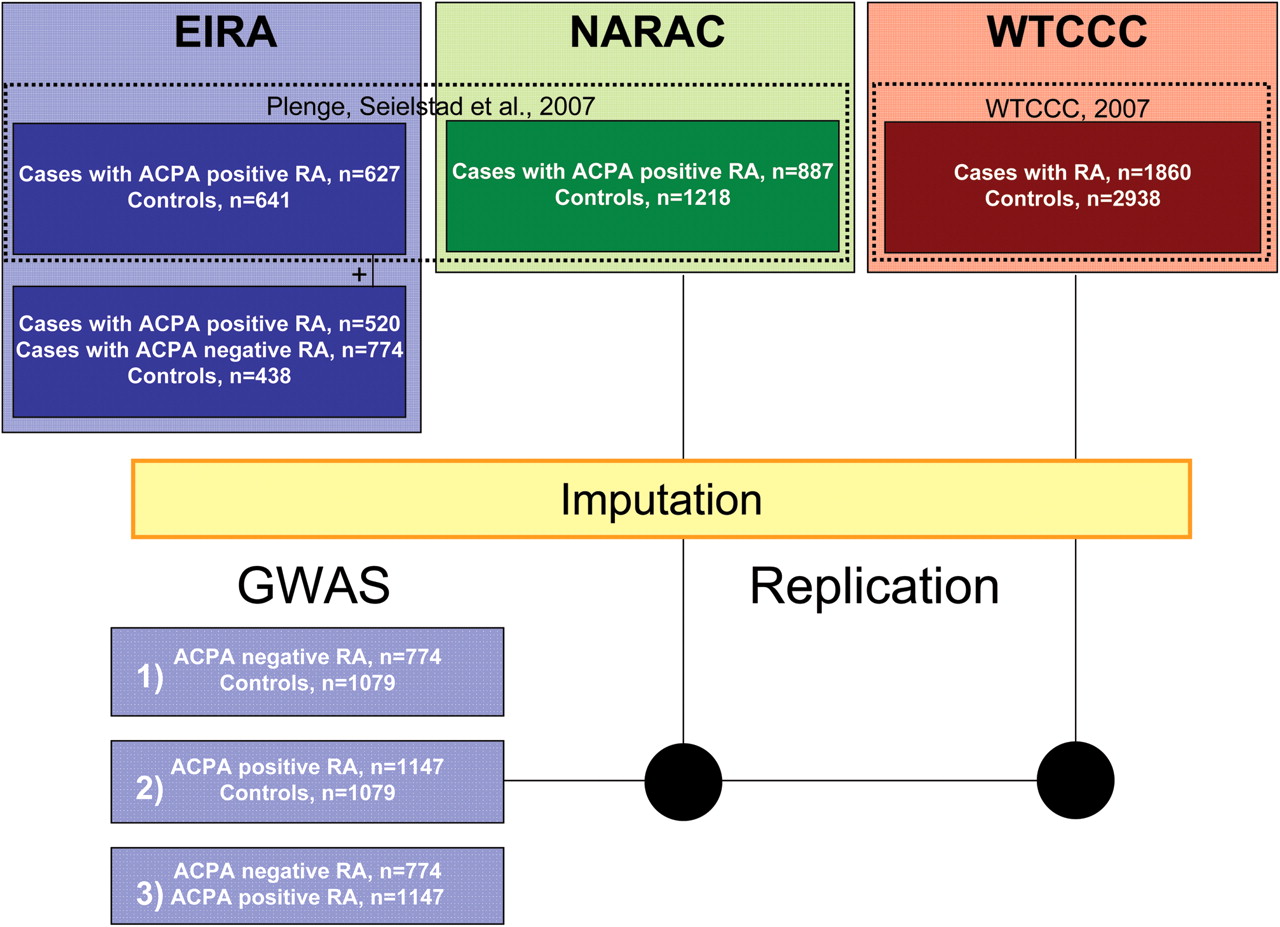
EIRA is a population-based case–control study enrolling incident (predominantly <1 year after clinical onset) cases of RA. The study base comprises residents aged 18–70 years in a geographically-defined area in the central and southern parts of Sweden. Details of the study design have been reported elsewhere.8 29 30 For each case a control was randomly selected from the study base by matching age, sex and residential area. For the present study we selected 3176 individuals (829 ACPA-negative cases of RA, 1218 ACPA-positive cases of RA and 1129 controls) for genome-wide genotyping. Informed consent was obtained from all participants and the ethical review board at the Karolinska Institutet approved the study. A portion of the data was included in a previously published GWAS of ACPA-positive RA13 (see table 1 in online supplement), but here we enlarge the GWAS dataset for ACPA-positive patients with RA and controls and include genome-wide data on ACPA-negative patients for the first time.
NARAC provided genotypes for 889 ACPA-positive patients and 1232 controls. The patients of self-reported white ancestry were recruited as prevalent (69.6%) or incident RA cases from several sites throughout North America. Nearly half of the cases (51.1%) reported a positive family history.13 Control subjects were selected on the basis of similar self-reported ancestry from 20 000 persons who were part of the New York Cancer Project. Written informed consent was obtained from all subjects who provided blood samples in accordance with protocols approved by the local institutional review boards.
The British RA population comprised 1860 patients and 3000 controls. Recruitment procedures have been described previously, together with frequencies of genetic variations.28
Demographic characteristics of patients and controls and the study logistics are shown in table 1 in the online supplement and in figure 1 where corresponding numbers after quality control procedures (see below) are shown. Genotyping, serological analysis and statistical evaluation are shown in the online supplement.

Work flow for genome-wide analysis (GWAS) of two subgroups of rheumatoid arthritis (RA). Three different Caucasian study populations represented in different colours (blue, green and red: Swedish, US, UK). Selection of data from previously published studies indicated by dashed rectangles with subsequent reference. Difference in sums is due to QC procedures at each stage of analysis. ACPA, antibodies to citrullinated peptide antigens.
Characteristics of the top associated SNPs from GWAS of ACPA-negative RA versus controls
Results
GWAS of ACPA-negative RA
We conducted a GWAS for ACPA-negative patients with RA with 774 cases and 1079 controls selected from the EIRA study. Both genotyped and imputed single nucleotide polymorphisms (SNPs) were included in the analysis. No single SNP reached genome-wide significance (figure 2). A Q-Q plot for observed versus expected p values is shown in figure 3A and shows no significant deviation from the expected distribution. As shown in table 1, five SNPs from three genetic loci had p values <10−5. One out of five was not associated with ACPA-positive RA while the other four (from two independent loci at chromosome 7) had only nominal association. These results indicate little or no overlap between the two RA subgroups for the five tentative SNPs that may associate with ACPA-negative RA in this GWAS.
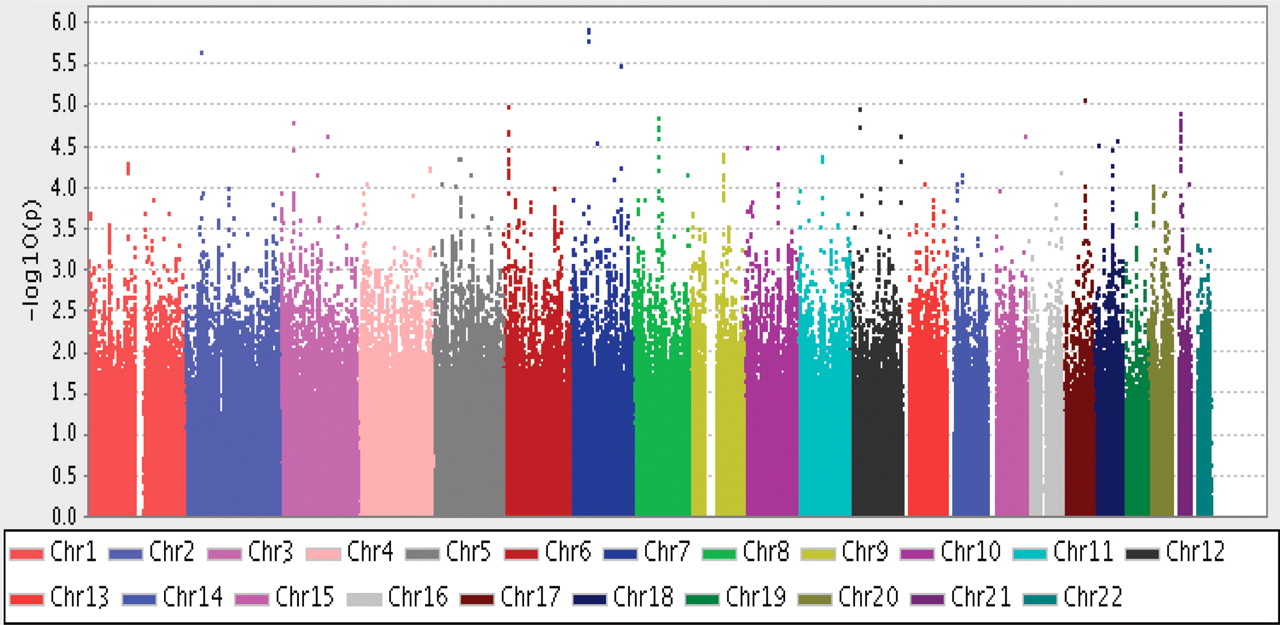
Probability plot for association with ACPA-negative rheumatoid arthritis (774 cases) versus 1079 controls, λGC = 1.0132 based on 1 723 056 single nucleotide polymorphisms. ACPA, antibodies to citrullinated peptide antigens.
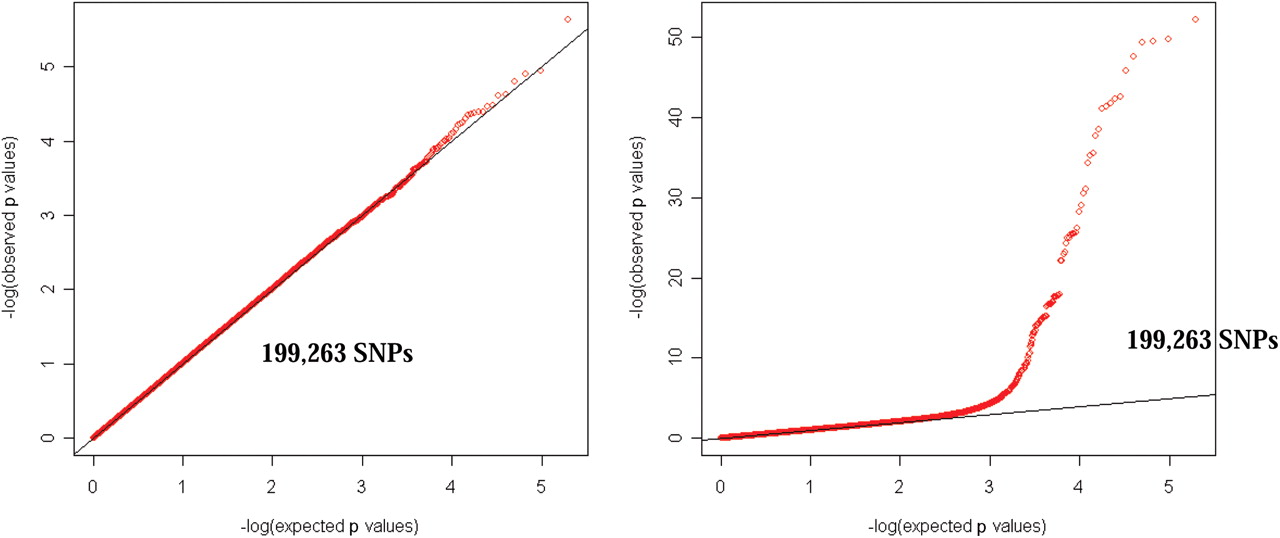
Quintile-quintile probability plots for observed versus expected distribution of p values for χ2 statistics for (A) ACPA-negative RA versus controls and (B) ACPA-positive RA versus controls. ACPA, antibodies to citrullinated peptide antigens; RA, rheumatoid arthritis; SNP, single nucleotide polymorphism.
Owing to our inability to identify any additional appropriately-sized case–control studies of ACPA-negative RA, we have so far been unable to replicate our findings for ACPA-negative RA.
GWAS of ACPA-positive RA
We have previously reported GWAS data on ACPA-positive RA based on a fraction of the EIRA study in combination with NARAC.13 In addition to the previously used 627 ACPA-positive RA cases and 641 controls from Sweden, we now selected 520 new ACPA-positive RA cases and 438 controls from the EIRA study for extension of the GWAS for ACPA-positive RA. Because all cases and controls were taken from the same study population, we combined all EIRA samples into a single analysis which included both genotyped and imputed SNPs. The diagram of genome-wide association for this analysis is shown in figures 4 and 5 and Q-Q plots are shown in figure 3B. Out of 1 723 056 analysed SNPs, we found 719 SNPs which passed a genome-wide significance threshold (see table 2 in online supplement). Of note, 718 of these SNPs were located within the HLA locus at chromosome 6 with physical positions between 31 279 236 and 33 164 413 (1 885 177 bp). A single non-HLA SNP rs2476601 was from PTPN22 gene at chromosome 1 with OR 1.66 (95% CI 1.40 to 1.96, p=9.77E-09). After relaxing the threshold for significance up to 10−6, an additional 196 SNPs were found to be significant from the HLA locus and seven non-HLA SNPs from the cluster of a ‘gene desert’ at chromosome 13 (see table 2 in online supplement). These seven SNPs are all in a recombination block according to HapMap data.31

Probability plot for association with ACPA-positive rheumatoid arthritis (1147 cases) versus 1079 controls, λGC = 1.0263 based on 1 723 056 single nucleotide polymorphisms (SNPs). Upper figure represents data for all SNPs including MHC locus and lower figure is without data for SNPs from MHC locus. ACPA, antibodies to citrullinated peptide antigens.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Probability plot for association for ACPA-positive RA (1147 cases) versus ACPA-negative RA (774 cases). λGC = 1.0029 based on 1 723 056 single nucleotide polymorphisms. ACPA, antibodies to citrullinated peptide antigens; RA, rheumatoid arthritis.
Characteristics of the top non-HLA SNPs from GWAS of ACPA-positive versus ACPA-negative RA for EIRA in comparison with NARAC and WTCCC studies
We subsequently extended the observations on patients with ACPA-positive RA in EIRA with a replication in NARAC using patients with ACPA-positive RA as cases and healthy individuals from a New York cancer surveillance study as controls. Many of the SNPs in the HLA region and one PTPN22 SNP (rs2476601) from EIRA were well replicated in NARAC (see table 3 in online supplement).
Frequencies of previously detected genetic variants for RA: comparison of meta-analysis of ACPA-positive RA patients from three populations with ACPA-negative RA patients
We also made an extra validation against the WTCCC study where, however, we were not able to separate patients based on ACPA status. It is known, however, that the large majority of the WTCCC RA cohort is rheumatoid factor (RF)-positive and, owing to a high correlation between RF and ACPA status, it most likely dominated by ACPA-positive RA cases.28 Many of the SNPs in the HLA region and one PTPN22 SNP (rs2476601) were well replicated also in WTCCC (table 3 in online supplement) with an overall OR for rs2476601in the three studies of 1.74 (95% CI 1.59 to 1.90, p=2.73045E-36, Mantel–Haenszel χ2 test for 17 520 chromosomes).
Contrasts between ACPA-positive and ACPA-negative RA
To formally test the hypothesis of a contrast between the two disease subgroups, we used the EIRA study to perform a direct comparison between ACPA-positive and ACPA-negative RA using the full GWAS data sets for these two populations of RA patients. The threshold for genome-wide significance was estimated as 2.9 × 10−8 (after Bonferroni correction for 1 723 056 tests). After corrections for multiple testing we found significant differences only in the HLA region of chromosome 6p with 814 SNPs spanning between 31 278 893 and 33 164 413 (see table 4 in online supplement). When we increased the threshold to 10−5 we identified an additional 352 SNPs in the HLA region targeted to physical positions in the extended HLA locus between 30 155 944 and 33 886 942 (3 730 998 bp) and three additional non-HLA SNPs (table 2): rs4305317 from chromosome 2 close to the LDHAL3 (lactate dehydrogenase A-like 3) gene, rs6448119 from chromosome 4 between the KCNIP4 (Kv channel interacting protein 4) and the GPR125 (G protein-coupled receptor 125) genes, and rs2961663 from chromosome 5 in the GMCL1L (germ cell-less homolog 1) gene.
The described differences in the HLA region between ACPA-positive and ACPA-negative disease are well in line with a previously published dataset on ACPA-positive and ACPA-negative patients with RA compared with controls which was focused only on the HLA using another set of SNPs together with classical PCR-based HLA-typing.7 32
Since the difference in allelic frequency in this analysis could be linked to the effect from any of the RA subgroups although it is dominated by susceptibility risk alleles for ACPA-positive RA, it is interesting to note that the two non-HLA SNPs rs4305317 and rs6448119 seem to be in association primarily with ACPA-negative RA.
Discussion
We report the first GWAS in one of the major subsets of RA defined by absence of ACPA reactivity, and the first comparison of GWAS results between ACPA-positive and ACPA-negative RA. Overall, we found significant differences in genetic associations between the two subsets, both concerning genes already described as being associated with ACPA-positive RA in previous genome-wide studies and concerning tentative associations that are preferentially seen for the ACPA-negative subset of RA. Notably, only very few genetic variations were associated with both RA subsets and those had a very minor influence on the genetic risk of RA.
The contrasting genetic aetiologies of ACPA-positive and ACPA-negative RA are most starkly evident at the HLA locus, and these data are in line with previous studies that were specifically focused on the HLA region and used other SNPs and smaller numbers than in the present study.7 32 These data therefore show that the functional conclusions concerning molecular pathogenesis of arthritis that have since long been deduced from the association with HLA genes, and particularly with variations in HLA class II genes, are valid only for the ACPA-positive subset of the disease; the implications of this discrepancy have also been discussed previously on the basis of data from classical PCR-based HLA genotyping.7 9 25 32
Concerning the non-HLA genes, several studies have now been published describing variations associated with either ACPA-positive RA13 14 17 27 or with RA where no discrimination between the two subsets was made.28 When analysing the published genetic variations that showed genome-wide or suggestive associations with ACPA-positive disease or with the entire RA population and also with the ACPA-negative RA subset in the EIRA study, the genetic variants in association with ACPA-positive RA were most often not significant for ACPA-negative RA and vice versa.
We performed an analysis of 14 well-established variations for association with ACPA-positive RA in combined material from Sweden, the USA and the UK and for ACPA-negative RA in the Swedish EIRA cohort (table 3). As can be seen from this analysis, most previously detected genetic variations (9/14) were associated only with ACPA-positive RA, three appeared to be associated with both subgroups, one appeared to be specific for ACPA-negative disease and one appeared to provide opposite results in the two subgroups. Since most efforts in the genetics of RA have been devoted to ACPA-positive RA, this difference may be due to selection rather than to a true absence of association with ACPA-negative RA. A recent twin study of ACPA-negative RA indicated that this phenotype is also genetically predetermined.33 Thus, our present negative results have to be interpreted with caution given the limited power of our investigations. Nevertheless, we can be confident that the major discrepancies described by GWAS—that is, the differential associations in the HLA region—are indeed true differences between the ACPA-positive and ACPA-negative RA subsets.
Concerning the non-HLA genes associated with the different subsets of RA, we performed additional genotyping to confirm the accuracy of imputation of SNPs using real genotyping data, when possible, to evaluate differences between the two RA subsets in order to decrease the risk of false positive findings due to use of imputations. Using this methodology we were not able to identify any strong genetic risk factors for ACPA-negative RA, while a number of SNPs from the HLA region as well as non-HLA SNPs were in association with ACPA-positive disease, confirming previously published data for ACPA-positive RA.13 28 Thus, although we cannot totally exclude overlapping genetic risk factors for the two RA subgroups, it is unlikely that this overlap is very big. We can also confirm the major differences between ACPA-positive and ACPA-negative RA concerning linkage to the HLA region.
Deciphering the pathogenesis of ACPA-negative RA remains a major challenge for genetic studies. In this study we found suggestive evidence—but not genome-wide significance-based evidence—for two new candidate loci in ACPA-negative disease (ie, RPS12P4 and IGFBP1). We also found suggestive evidence for the previously described IRF5 locus as susceptibility genes for ACPA-negative RA. Thus, most of the difference in genetic factors between ACPA-negative and ACPA-positive RA is seen in the HLA region, close to the HLA-DRB1 locus. The non-HLA variant rs4305317, close to RPS12P4 at chromosome 2, is the best candidate for association with ACPA-negative RA but not with ACPA-positive RA. It should be emphasised, however, that this association as well as other data related to associations with ACPA-negative RA need independent replication owing to the limited size of the present study.
GWAS provide new potentials to determine genetic variations and molecular pathways that are shared between different inflammatory diseases. The associations of several diseases and disease subsets with PTPN22, CD40, TRAF1-C5 and STAT4 are typical examples of this sharing.17 27 34 35 The present study illustrates the other complementary perspective—namely, the potential also to use GWAS to subdivide criterion-based diseases such as RA into new entities. As exemplified here, one subset of a disease may then share certain risk genes and possibly pathogenic pathways with some other autoimmune entities, as is the case for PTPN22 and ACPA-positive RA and type I diabetes,36 whereas the other subset of the same criterion-based disease may share genetic linkages and pathogenetic pathways with still other autoimmune entities. We can expect refined classifications of criterion-based diseases such as RA to lead to the use of genetics to provide an indication of what molecular pathways are involved in the pathogenesis of different subsets of today's criterion-based diseases. This knowledge should, in turn, be indispensable when trying to find treatments to target these pathways in patients in these different and distinct subsets of chronic autoimmune diseases.
Acknowledgments
The authors acknowledge the help of Peter K Gregersen and Jane Worthington in access to NARAC and WTCCC data. They also thank Robert M Plenge for careful reading of the manuscript and critical discussions; Kian Mun Chan, Boon Yeong Goh, Wee Yang Meah, Jameelah B S Mohamed, Jason Ong, Eileen Ping and Sigeeta Rajaram for their invaluable laboratory assistance; the participating patients with RA and controls and all rheumatologists for recruiting patients in the EIRA study; and Marie-Louise Serra, Camilla Bengtsson, Eva Jemseby and Lena Nise for their invaluable contributions to the collection of data and maintenance of the database.
References
Supplementary materials
Web Only Data
Files in this Data Supplement:
Footnotes
LP and MS contributed equally to this work.
-
Competing interests None.
-
Funding Supported by grants from the Swedish Research Council, the Swedish Council for Working Life and Social Research, King Gustaf V's 80-year Foundation, the Swedish Rheumatism Foundation, the insurer, AFA and the EU-supported projects AutoCure and Masterswitch. The Agency for Science Technology and Research (ASTAR), Singapore supported the genotyping and data analysis. The funding agencies had no influence on study design, evaluation of results and decision to publish the data.
-
Ethics approval This study was conducted with the approval of the Regional Ethical Review Board in Stockholm.
-
Provenance and peer review Not commissioned; externally peer reviewed.