Article Text

Abstract
Background After adalimumab treatment failure, tumour necrosis factor inhibition (TNFi) and non-TNFi biological disease-modifying anti-rheumatic drugs (bDMARDs) are equally viable options on a group level as subsequent treatment in rheumatoid arthritis (RA) based on the current best evidence synthesis. However, preliminary data suggest that anti-adalimumab antibodies (anti-drug antibodies, ADA) and adalimumab serum levels (ADL) during treatment predict response to a TNFi as subsequent treatment.
Objective To validate the association of presence of ADA and/or low ADL with response to a subsequent TNFi bDMARD or non-TNFi bDMARD. Sub-analyses were performed for primary and secondary non-responders.
Methods A diagnostic test accuracy retrospective cohort study was done in consenting RA patients who discontinued adalimumab after >3 months of treatment due to inefficacy and started another bDMARD. Inclusion criteria included the availability of (random timed) serum samples between ≥8 weeks after start and ≤2 weeks after discontinuation of adalimumab, and clinical outcome measurements Disease Activity Score in 28 joints - C-reactive protein (DAS28-CRP) between 3 to 6 months after treatment switch. Test characteristics for EULAR (European League Against Rheumatism) good response (DAS28-CRP based) after treatment with the next (non-)TNFi bDMARD were assessed using area under the receiver operating characteristic and sensitivity/specificity.
Results 137 patients were included. ADA presence was not predictive for response in switchers to a TNFi (sensitivity/specificity 18%/75%) or a non-TNFi (sensitivity/specificity 33%/70%). The same was true for ADL levels in patients that switched to a TNFi (sensitivity/specificity 50%/52%) and patients that switched to a non-TNFi (sensitivity/specificity 32%/69%). Predictive value of ADA and ADL were similar for both primary and secondary non-responders to adalimumab.
Conclusions In contrast to earlier research, we could not find predictive value for response to a second TNFi or non-TNFi for either ADA or random timed ADL.
- rheumatoid arthritis
- anti-TNF
- DMARDs (biologic)
- DAS28
- disease activity
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Anti-adalimumab antibody (anti-drug antibody, ADA) presence has been suggested to correlate with response to a second biological disease-modifying anti-rheumatic drug (bDMARD) after discontinuation of adalimumab use.
What does this study add?
We investigated the predictive value of ADA and adalimumab serum levels (ADL) for EULAR (European League Against Rheumatism) clinical response to subsequent treatment with a second bDMARD (tumour necrosis factor inhibition (TNFi) or non-TNFi) after discontinuing adalimumab because of treatment failure.
How might this impact on clinical practice or future developments?
Neither ADA presence nor ADL had predictive value for clinical response to a subsequent TNFi or non-TNFi treatment after failure of adalimumab treatment.
Combining these data with four earlier studies that did find some predictive value of adalimumab and etanercept (anti)drug levels, the next research step might be doing a well-dimensioned prospective trial.
Introduction
Biological disease-modifying anti-rheumatic drugs (bDMARDs) are important in the treatment of rheumatoid arthritis (RA). bDMARDs with several modes of action are available, such as tumour necrosis factor inhibition (TNFi) (eg, adalimumab, etanercept, golimumab, infliximab, certolizumab) and non-TNFi (eg, rituximab, tocilizumab, abatacept). Adalimumab—a human monoclonal antibody TNFi—is one of the most frequently used bDMARDs, and is a safe and effective treatment for RA.
However, approximately 41% of RA patients do not achieve good response1–3 after 6 months of treatment with adalimumab.4 5 After non-response to adalimumab (or any bDMARD) treatment, current guidelines state that another TNFi or a non-TNFi bDMARD could be prescribed as subsequent treatment with equal chance of response.6 This is supported by current available evidence from four randomised controlled trials (RCT’s),7–10 and two systematic reviews on predictive factors for response to a bDMARD in RA.11 12 Based on this, no preference should be given to starting either another TNFi, or a non-TNFi bDMARD after primary or secondary non-response to adalimumab.13
However, it has been suggested that measurement of adalimumab serum levels and/or anti-adalimumab antibodies (therapeutic drug monitoring, TDM) might be helpful for channelling the right patients to a TNFi or a non-TNFi thus increasing overall response chances.14–16 The rationale for this is that approximately 20% to 50% of the RA patients treated with adalimumab develop antibodies against this drug (anti-drug antibodies, ADA) and this can result in primary or secondary non-response.14–16 Another possible reason for non-response, however, is innate insensitivity to TNFi in a proportion of patients. It can be hypothesised that the first group of non-responders will have adequate response chances to a second TNFi, whereas in the second group of patients, TNFi response will be much lower. This is supported by several cohort studies and a recent systematic review in RA and axial spondylarthritis, for adalimumab and infliximab.14–17
Following this rationale, the optimal strategy after adalimumab non-response might be a second TNFi in patients with low adalimumab levels/ADA presence, and a non-TNFi in patients with adequate levels and no ADA presence. One could argue that just giving a non-TNFi in all adalimumab non-responders would negate the need for testing. However, many adalimumab non-responding patients experience secondary non-response rather than primary non-response, and patients in which secondary non-response occurred were indeed TNFi responding patients. Therefore, response rates to a second TNFi in these patients might be higher than response rates to a non-TNFi, resulting a better outcome for all patients after TDM.
The above-mentioned hypothesis has—in part—been tested in two studies with infliximab and adalimumab.15 18 These studies showed that presence of ADA against either infliximab or adalimumab was associated with a larger decrease in disease activity after the next TNFi. Additionally, the same mechanism has been replicated using infliximab in RA, and adalimumab in axial spondyloarthritis.14 16 However, these studies have some limitations; the number of patients was somewhat limited, and no differentiation was made between primary and secondary non-responders, a distinction that might be important for response chances to a second TNFi, as argued earlier. Also, these studies did not mention test characteristics (sensitivity/specificity), only difference in mean improvement, thus hampering judgement of test characteristics. In addition, the studies did not assess the predictive value of adalimumab TDM for response to non-TNFi after adalimumab, which is relevant to determine whether ADA presence is simply a marker of more refractory disease or able to differentially predict response to a second TNFi compared with a non-TNFi . Finally, testing with a newer competitive ELISA is now possible in order to quantify anti-drug antibodies even in the presence of large amounts of TNF inhibitor.19 As this is a drug-tolerant assay, it is a more precise measure of ADA than conventional testing methods where ADA cannot be detected in the presence of large amounts of the drug.
Therefore, we set out to investigate this predictive value in a larger study population, estimating sensitivity and specificity of both presence of ADA and random timed adalimumab levels (ADL), and validate currently proposed thresholds, in both patients that switched to a TNFi (TNFi switchers) and patients that switched to a non-TNFi (non-TNFi switchers).
Methods
Design
A retrospective diagnostic test accuracy cohort study to assess the predictive value of ADA and ADL for response to a subsequent TNFi or non-TNFi bDMARD in RA patients.
Patients
All RA patients who received adalimumab and subsequently another TNFi (etanercept, golimumab, infliximab, certolizumab) or a non-TNFi bDMARD (rituximab, tocilizumab, abatacept) in the Sint Maartenskliniek or Radboud University Medical Centre (Radboudumc) between January 2012 and January 2018 were considered for inclusion in the current study. Potentially eligible participants were identified through the electronic patient records of the Sint Maartenskliniek and the Radboudumc. Patients included in this study had a diagnosis of RA according to American College of Rheumatology (ACR) (1987) or ACR/European League Against Rheumatism (EULAR) (2010) criteria,20 21 or clinical diagnosis, and were ≥16 years of age. They had received adalimumab for at least 3 months (+/−2 weeks) in standard dosing (40 mg subcutaneously every other week). Acceptable reasons for stopping adalimumab were either inefficacy (primary or secondary, no formal disease activity cut-off) or toxicity, but not tapering because of remission. The next bDMARD should also have been administered in standard dosing (registered dose, exception being rituximab 1×1000/2×500 mg instead of 2×1000 mg) for at least 3 months (+/−2 weeks). Furthermore, a serum sample that is suitable for analysis should be available, being samples taken ≥8 weeks after start adalimumab and within 2 weeks after discontinuing adalimumab (for ADL) or within 12 weeks after discontinuation (for ADA).22 Serum samples were derived from a period of biobanking at every visit of RA patients and an observational cohort study including consecutive bDMARD starters. Finally, Disease Activity Score in 28 joints - C-reactive protein/erythrocyte sedimentation rate (DAS28-CRP/ESR) scores had to be available to assess EULAR clinical response to subsequent bDMARDs, a baseline DAS at start and a follow-up DAS after 3 to 6 months of treatment (+/−8 weeks).
Ethical approval, consent and funding
Approval from the local ethics committee (Commissie Mensgebonden Onderzoek (CMO) region Arnhem-Nijmegen) was obtained (CMO: 2019–5443). Patients had either previously consented to inclusion in several biobanking studies, including the Nijmegen RA protocollaire follow-up23 (CMO-number: 2016–2281) and the BIOTOP study24 (CMO region Arnhem-Nijmegen, NL47946.091.14) or were sent opt-out informed consent letters with information about the aims and methods of the study. Patients were given 4 weeks to read the information and respond in case they are not willing to participate (according to Dutch law: WGBO art 458 sub 2). This study received no external funding. The laboratory analyses of adalimumab and ADA levels and personnel costs were funded by the Sint Maartenskliniek.
The study was conducted according to the principles of the Declaration of Helsinki and in accordance to Dutch law: WMO, AVG, WGBO, code Goed Gedrag and NFU ‘richtlijn kwaliteitsborging mensgebonden onderzoek’.
Testing of serum adalimumab levels and anti-adalimumab antibodies
Blood samples were pseudonymised and stored at −80°C in the Sint Maartenskliniek or the Radboudumc biobank for collection. A drug-tolerant competitive enzyme-linked immunosorbent assay (Sanquin, the Netherlands) was used to quantify ADA, enabling measurement of ADA in the presence of large amounts of TNF-inhibitor. In short, a high affinity adalimumab mutant (variant cb1-3, murine origin25) was used, which efficiently removes the TNFi from TNF due to increased affinity.
Thereafter, the adalimumab concentration was determined via an ELISA. Concentrations <0.004 µg/mL were deemed not detectable. Concentrations <5 µg/mL were considered as not effective.26 27 ADA were quantified with the antigen binding test (radioimmunoassay), with a reference value of >12 AU/mL.14 15
Testing was performed by Sanquin, The Netherlands. The treating physician (who was responsible for the choice of subsequent bDMARD) was blinded to test results as sample analysis was done retrospectively.
Assessment of clinical outcome
The primary outcome of this study was the association between ADA or ADL and EULAR good response to the bDMARD after adalimumab failure (‘EULAR response’). Response was operationalised as EULAR good response to the subsequent bDMARD after adalimumab failure, measured between 3 to 6 months (+/−8 weeks) after start of the next and subsequent bDMARDs based on the DAS28-CRP/ESR, which is a valid, reliable and broadly accepted indicator of the clinical activity of RA.2 3 When DAS28 response was unavailable/if glucocorticoid injection could have influenced the DAS28 score outcome, clinical assessment by a rheumatologist was used to assess response (‘clinical response’). When DAS28 was low at baseline and remained low in follow-up, the clinical response assessment was also used. Of note, both DAS28-ESR-scores and DAS28-CRP scores were used during the study period, and slightly different cut-offs for response were used to change from baseline >=1.2 and current DAS28-ESR <3.2 and DAS28-CRP <2.9, respectively, to consistently assess response.28
Finally, a subanalysis was performed for primary and secondary failure on adalimumab. Non-response is classified as primary non-response if adalimumab was used for less than 12 months and patients had not shown clinical response at any point, and as secondary non-response if adalimumab is used for longer than 12 months or if patients had at any point shown clinical response. No fixed disease activity cut-off was used for this classification due to the retrospective nature of the study.
Statistical analyses
Data management systems Castor EDC and Microsoft Power BI database were used to enter and store the data. Data was extracted to a Stata database and analysed (V.13.1).
Descriptive statistics are provided with mean (+/−SD), median (IQRs (p25 to p75)) or n (%) depending on data distribution. Baseline characteristics of the TNFi vs non-TNFi as second treatment groups were compared using a Student’s t-test (or, if not normally distributed, Wilcoxon rank-sum) and χ2 test for continuous and categorical data, respectively.
Correlations between ADA presence and clinical variables (ie, age, gender, smoking, disease duration, rheumatoid factor, anti-citrullinated protein antibodies, DAS28-CRP/ESR and its’ components, CRP and ESR) were first cross-sectionally explored by Spearman’s correlation analysis.
Area under the receiver operating characteristic (AUROC) curves were generated to evaluate the predictive value of ADA presence and ADL for EULAR clinical response in respectively TNFi and non-TNFi as consecutive treatment. Sensitivity and specificity were calculated using the cut-offs suggested by earlier studies (ADL <5 mg/L26 27 and ADA >12 AU/mL14 15), and precision is shown with a 95% CI. A p value <0.05 was considered statistically significant.
Results
Participants
One hundred and thirty-seven patients were included, of which 93.4% met the 1987 ACR or 2010 ACR/EULAR criteria (exclusion of the nine patients that did not meet either criteria did not significantly alter the results). Forty-seven of the 137 patients switched to a second TNFi and 90 to a bDMARD with another mode of action (figure 1). ADA were measured in all patients and ADL were measured in 92 patients due to timing of serum samples. Baseline characteristics and group differences are shown in table 1. The sample populations have comparable baseline values.
Twelve patients were assessed by means of DAS28-ESR. One hundred and two patients were assessed by means of DAS28-CRP. The remaining 23 patients were assessed by clinical response, in about one-fourth of these cases this was needed because baseline was DAS28-ESR and follow-up was DAS28-CRP or vice versa and no valid cut-off could be used to assess response. The remaining patients (three-fourth) were assessed by clinical response due to missing a DAS28 measure.
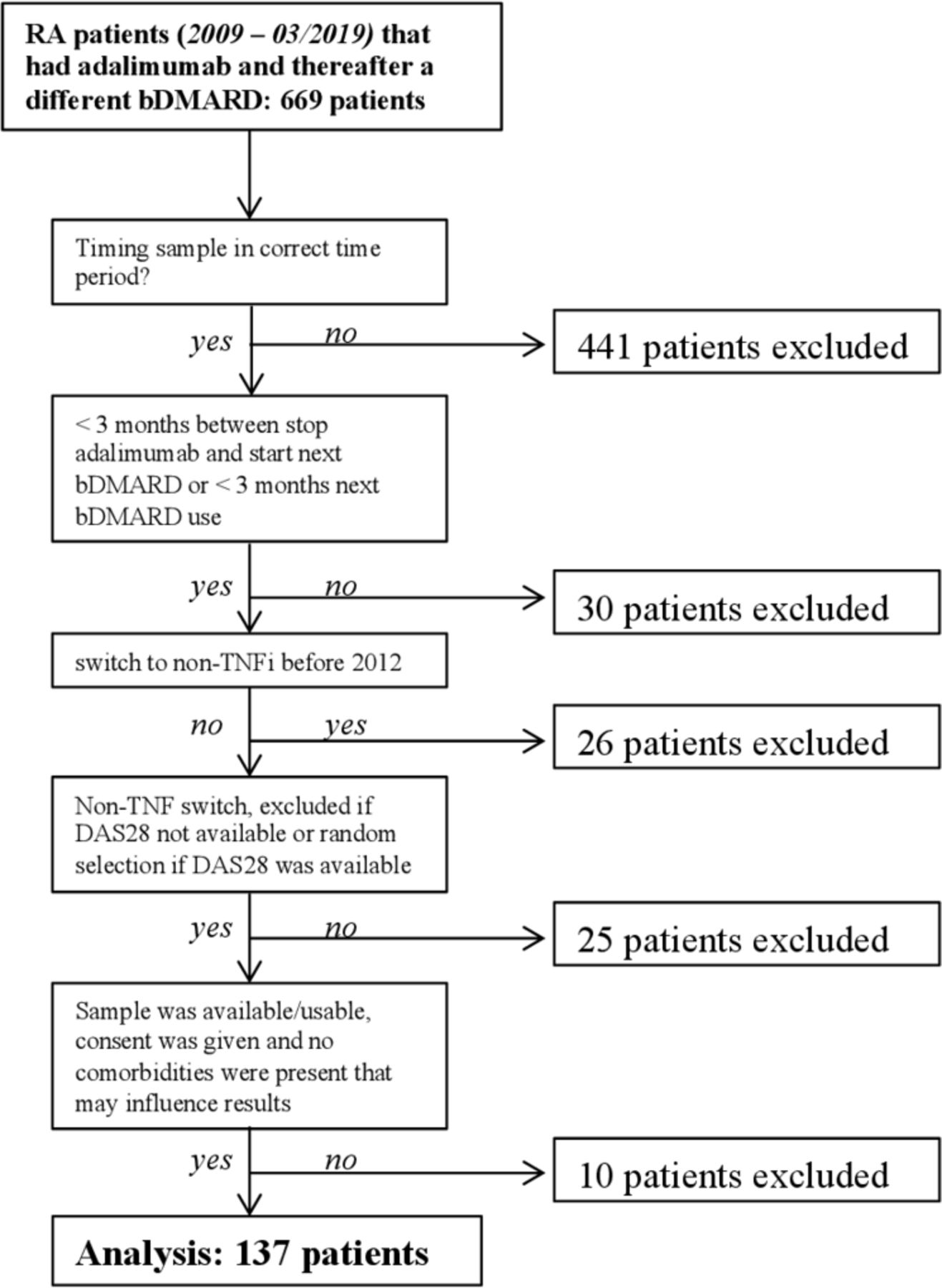
Flow of participants. bDMARD, biological disease-modifying anti-rheumatic drug;DAS28, Disease Activity Score in 28 joints; RA, rheumatoidarthritis; TNFi, tumour necrosis factor inhibition.
Baseline values and differences between groups
In patients receiving a second TNFi, 36% achieved good EULAR clinical response, while 23.4% achieved good EULAR clinical response in the non-TNFi group. ADA were present in 39 of 137 patients. ADL >5 µg/mL in 35 of 92 patients. It is often hypothesised that co-treatment with a csDMARD reduces antibody formation. In our study, the percentage of patients was identical in both csDMARD treated and non-csDMARD treated at 28%.
Results of adalimumab levels were also similar, with 38% and 39% of csDMARD treated and not csDMARD treated patients, respectively, having adalimumab levels >5 µg/mL.
Correlations between ADA/ADL and patient characteristics
ADL showed a negative correlation with baseline DAS28 (Spearman’s ρ=−0.68, p=0.00). However, ADA presence did not correlate significantly with baseline DAS28 (ρ=0.23, p=0.28) and both ADA and ADL did not correlate with follow-up DAS28 (ρ=−0.29, p=0.17, and ρ=0.10, p=0.65 respectively). Absolute numbers for DAS28 and ADA/ADL as well as baseline ESR and CRP are added as supplementary tables.
ADA correlates with baseline ESR (ρ=0.49, p=0.01) and ADL with baseline CRP (ρ=–0.67, p=0.00) and ESR (ρ=–0.546, p=0.006).
Predictive value of ADA and ADL
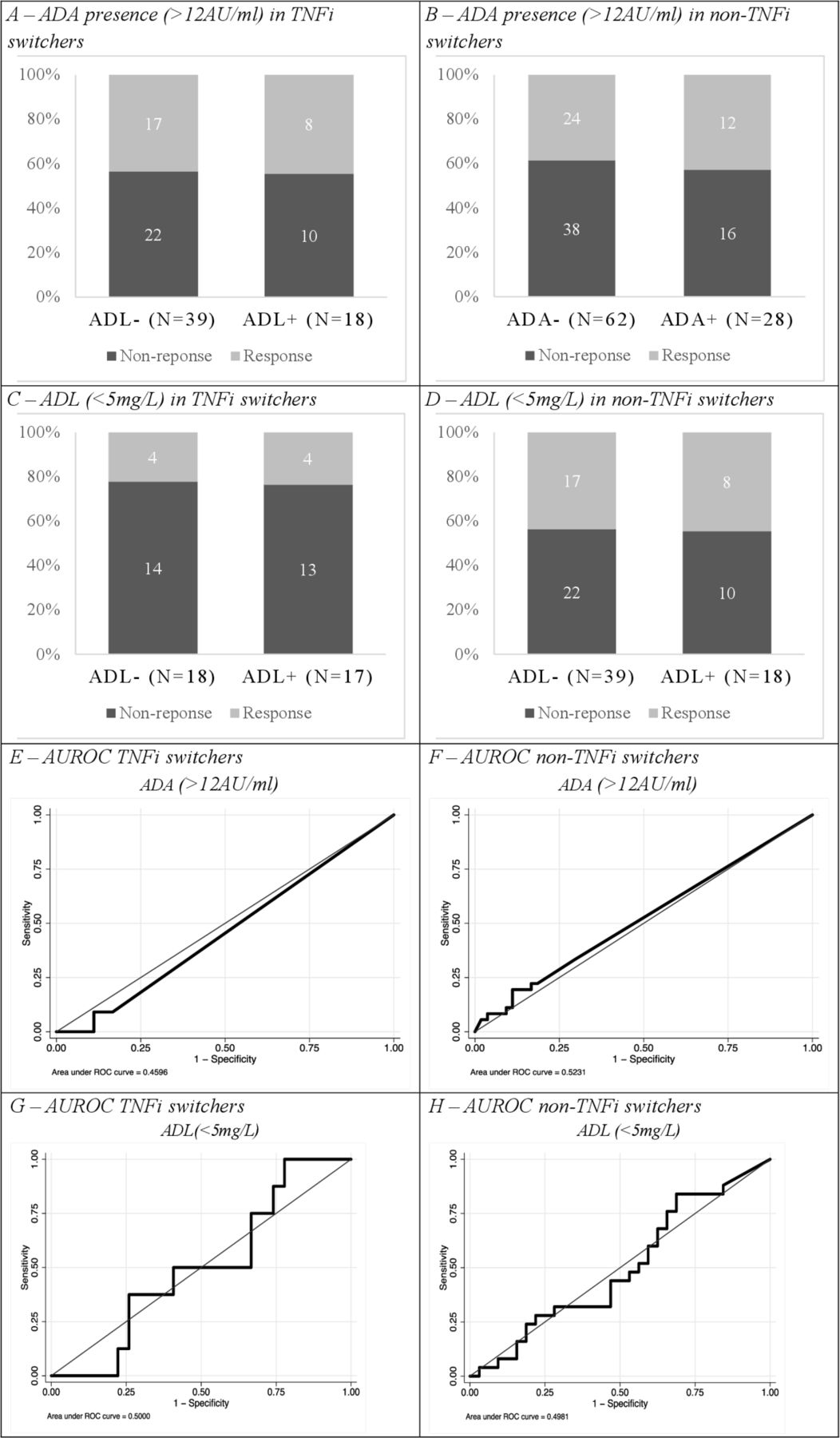
No clear predictive value of ADA could be found in either TNFi or non-TNFi groups (figure 2). In the TNFi switchers, a sensitivity of 18% and specificity of 75% were found for presence of ADA predicting EULAR good response, with an AUROC value 0.46 (95% CI=0.32 to 0.59). For non-TNFi switchers, a sensitivity of 33% and specificity of 70% were found and the AUROC value was 0.52 (95% CI=0.42 to 0.63).

{kind=link}
{kind=link}
Response and ADA presence in TNFi switchers (A) and non-TNFi switchers (B). Adalimumab levels <5 mg/L in TNFi switchers (C) and non-TNFi switchers (D). AUROC of ADA in TNFi switchers (E) and non-TNFi switchers (F). AUROC of ADL in TNFi switchers (G) and non-TNFi switchers (H). ADA, anti-drugantibodies; ADL, adalimumab; AUROC, area under thereceiver operating characteristic; TNFi, tumour necrosis factorinhibition.
Additionally, in respect to ADL levels no predictive value was observed in the TNFi or non-TNFi group. In the TNFi switchers a sensitivity of 32% and specificity of 69% were found for ADL predicting EULAR clinical response, with an AUROC value of 0.50 (95% CI=0.29 to 0.71), whereas in the non-TNFi switchers a sensitivity of 50% and specificity of 52% were found, with an AUROC value of 0.50 (95% CI=0.34 to 0.65).
Secondary outcomes
ROC analysis was conducted for patients with primary and secondary non-response as a mechanistic difference was expected between these groups. There were 53 patients with primary failure of which 45 had switched to a non-TNFi and 8 had switched to a TNFi. There were 84 patients with secondary failure of which 45 had switched to a non-TNFi and 39 had switched to a TNFi. Clinical response was not significantly different in the secondary failures than in the primary failures (32.1% vs 37.7% respectively, p=0.580). Additionally, there was no significant difference in ADA presence (26.2% vs 32.1%, p=0.560) or drug levels >5 µg/mL (32.5% vs 42.3% p=0.390) between the primary and secondary non-response groups.
ADA and ADL also did not show predictive value for response to either a second TNFi or a non-TNFi in subanalyses restricted to primary or secondary non-responders specifically (table 2).
Predictive values of ADA and ADL for primary and secondary non-responders in TNFi and non-TNFi switchers
Discussion
In this diagnostic test accuracy study, in contrast to other studies, no predictive value for response to a second (non-)TNFi was found for either ADA or random timed ADL.
This is due to the fact that sensitivity and specificity was assessed instead of mean DAS values. There were, however, some significant correlations were found as previously reported in other studies. Not only did the results of this study show no predictive values, in some analyses a prediction is found in the opposite direction of what was expected. The AUROC values were all close to 0.5 which shows that this is not likely due to lack of power.
This study has several strengths: First, the choice of treatment and outcome assessment were blinded for ADA/ADL as these had not been determined at time of treatment. Second, a larger patient sample was achieved than in previous studies, except for the 2019 L‘Ami study.29 Third, there was solely focussed on adalimumab. Fourth, selection bias is unlikely because the inclusion criteria for the several studies in which patients were included when their sample was drawn were very broad. Finally, a control group was included with patients that switched to a non-TNFi treatment, to assess whether any predictive value of ADA/ADL was different for a second TNFi versus a non-TNFi bDMARD, as otherwise ADA/ADL might simply predict a more severe disease phenotype instead of differing chances of response to TNFi versus non-TNFi.
However, several limitations of this study should also be addressed. First, the samples were not taken at through level but rather timed at random related to adalimumab injection. This might have reduced the association between (anti)drug levels and response. However, it should be noted that random timed drug levels, and moreover ADA, are strongly correlated with trough level sampling.30 So this should not have resulted in absence of any predictive value. In addition, random timed drug sampling is more feasible in clinical practice, thus increasing generalisability.
Second, as this was a retrospective study, both serum samples and clinical outcomes were not always available, and this might have resulted in selection bias.
Finally, misclassification of the outcome can occur, both by incorrectly classifying patients as responders (eg, by glucocorticoid injections resulting in spuriously low DAS28) or incorrectly classifying as non-responder (eg, a patient starting with a low baseline DAS28 that remains low during treatment). To correct this misclassification, the physician judgement of response was also assessed in a sensitivity analysis, which did not lead to a different result.
Further research should confirm if ADA presence and ADL are indeed not predictive for disease activity. This could be done in a prospective study with a large sample size in which DAS28 measurements and sample collection are done on the correct time points in all patients. Ideally, disease activity should always be assessed with use of validated scores, not physician judgement for disease activity. An RCT to address these is currently evaluating whether a switching strategy based on ADL is superior to usual care switching in RA patients failing adalimumab treatment.
Whilecounterintuitive, it is hard to find an explanation for the lack of a positive finding. It should be noted that this is also true for a very related other adalimumab TDM issue; low ADL/ADA presence are not predictive for being able to successfully stop adalimumab use.17 The underlying reasoning is in fact the same; a drug should not work when levels are low/absent and/or ADA are present. This should be studied further, as it may offer new insights in the mode of action TNFi in general.
For now, a rheumatologists’ decision to switch to a TNFi/non-TNFi treatment after adalimumab failure should not be led by the idea that one could be more effective than the other. Therefore, rheumatologists should let their decision be led by other important variables such as possible side effects, local protocol, economical aspects and patient preferences.
Acknowledgments
The authors would like to thank all the patients who were willing to participate and Thea van Gaalen for helping with the collection of samples. We would also like to thank Bronke Boudewijns for tracking down data on short notice.
References
Footnotes
Handling editor Josef S Smolen
Contributors EU, NdB, AdB, NvH and BvdB were involved in the conception and design of the study. EU, NdB, AB, LT and IM were involved in the data collection. EU, NdB and AdB contributed to the data analysis. EU, NdB, MW and AdB drafted the manuscript and all co-authors reviewed the manuscript critically and gave final approval for its submission.
Funding This study was not supported by any external funding. The laboratory analyses of adalimumab and antibodies and serum levels (ADA and ADL) were performed in the laboratory of Sanquin, Amsterdam.
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting or dissemination plans of this research.
Patient consent for publication Not required.
Ethics approval Approval from the local ethics committee (Commissie Mensgebonden Onderzoek (CMO) region Arnhem-Nijmegen) was obtained (CMO: 2019–5443). Patients had either previously consented to inclusion in several biobanking studies, including the Nijmegen RA protocollaire follow-up23 (CMO-number: 2016–2281) and the BIOTOP study24 (CMO region Arnhem-Nijmegen, NL47946.091.14) or were sent opt-out informed consent letters with information about the aims and methods of the study. Patients were given 4 weeks to read the information and respond in case they are not willing to participate (according to Dutch law: WGBO art 458 sub 2). This study received no external funding. The laboratory analyses of adalimumab and ADA levels and personnel costs were funded by the Sint Maartenskliniek.
The study was conducted according to the principles of the Declaration of Helsinki and in accordance to Dutch law: WMO, AVG, WGBO, code Goed Gedrag and NFU ‘richtlijn kwaliteitsborging mensgebonden onderzoek’.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. Additional unpublished data can be obtained from the corresponding author upon reasonable request.