Article Text

Abstract
Objective The strong genetic contribution of the major histocompatibility complex (MHC) region to rheumatoid arthritis (RA) has been generally attributed to human leukocyte antigen (HLA)-DRB1. However, due to the high polymorphisms and linkage disequilibrium within MHC, it is difficult to define novel and/or independent genetic risks using conventional HLA genotyping or chip-based microarray technology. This study aimed to identify novel RA risk variants by performing deep sequencing for MHC.
Methods We first conducted target sequencing for the entire MHC region in 357 anticitrullinated protein antibodies (ACPA)-positive patients with RA and 1001 healthy controls, and then performed HLA typing in an independent case–control cohort consisting of 1415 samples for validation. All study subjects were Han Chinese. Genetic associations for RA susceptibility and severity were analysed. Comparative modelling was constructed to predict potential functions for the newly discovered RA association variants.
Results HLA-DQα1:160D conferred the strongest and independent susceptibility to ACPA-positive RA (p=6.16×10−36, OR=2.29). DRβ1:37N had an independent protective effect (p=5.81×10−16, OR=0.49). As predicted by comparative modelling, the negatively charged DQα1:160D stabilises the dimer of dimers, thus may lead to an increased T cell activation. The negatively charged DRβ1:37N encoding alleles preferentially bind with epitope P9 arginine, thus may result in a decreased RA susceptibility.
Conclusions We provide the first evidence that HLA-DQα1:160D, instead of HLA-DRB1*0405, is the strongest and independent genetic risk for ACPA-positive RA in Han Chinese. Our study also illustrates the value of deep sequencing for fine-mapping disease risk variants in the MHC region.
- rheumatoid arthritis
- gene polymorphism
- ant-ccp
Statistics from Altmetric.com
Key messages
What is already known about this subject?
The strong genetic contribution of the major histocompatibility complex (MHC) region to rheumatoid arthritis (RA) susceptibility has been generally attributed to HLA-DRB1.
However, due to the high linkage disequilibrium in MHC, it is difficult to define novel and/or independent RA risks using the conventional HLA genotyping or chip-based microarray technology.
What does this study add?
We first conducted target sequencing for the entire MHC region and then performed HLA typing in an independent cohort for validation.
We provide the first evidence that (1) HLA-DQα1:160D is the strongest and independent genetic risk for anticitrullinated protein antibodies (ACPA)-positive RA in Han Chinese, and (2) HLA-DRβ1:37N is a novel and independent protective factor for ACPA-positive RA.
We next performed a structure-based functional prediction for the two variants.
We showed that (1) negatively charged DQα1:160D (D160α) stabilised the structure of DQα1-DQβ1 dimer of dimers and may lead to an increased T cell activation, and (2) DRβ1:37N benefits electrostatic interaction and preferentially binds with epitope P9 arginine, thus may result in a decreased RA susceptibility.
Key messages
How might this impact on clinical practice or future developments?
A key finding of this study is the major genetic influence of HLA-DQα1:160D, instead of the well-known HLA-DRB1 alleles, on ACPA-positive RA in Han ancestry.
It may indicate that HLA-DQα1:160D could be a potential therapeutic target for RA.
Introduction
Rheumatoid arthritis (RA) is a chronic and systemic autoimmune disease primarily affecting the peripheral joints. Studies indicate that RA is a heterogeneous disease where different subsets result from complex interactions between genetic and environmental factors.1–3 The major histocompatibility complex (MHC) genes represent the best described genetic risk loci linking to RA susceptibility in many populations. HLA-DR variants were mainly associated with anticitrullinated protein antibodies (ACPA)-positive RA.4–7 Furthermore, the RA-risk human leukocyte antigen (HLA) alleles are heterogeneous among ethnic groups. For example, HLA-DRB1*04:01 is the major RA-risk allele in Caucasians, whereas DRB1*04:05 is the most frequent RA-risk allele in East Asians.5 8–10 In addition to HLA-DRB1, other HLA genes, such as HLA-B and HLA -DPB1, have also been suggested to play a role in susceptibility to RA.6 11 However, due to the high linkage disequilibrium (LD) in the MHC region and high-density genes within the MHC region, it is difficult to identify the ‘real’ and/or additional independent genetic risks by the conventional HLA genotyping and chip-based microarray technology.12 13
To fine-map the MHC region and identify novel variants contributing to RA susceptibility, we performed deep sequencing for the entire MHC region and HLA typing for validation. Given that a particular amino acid(s) rather than its position may have potential biological function(s), in the present study we have mainly focused on the individual amino acid variants, classical HLA alleles and single nucleotide polymorphism (SNPs).
Materials and methods
Study design
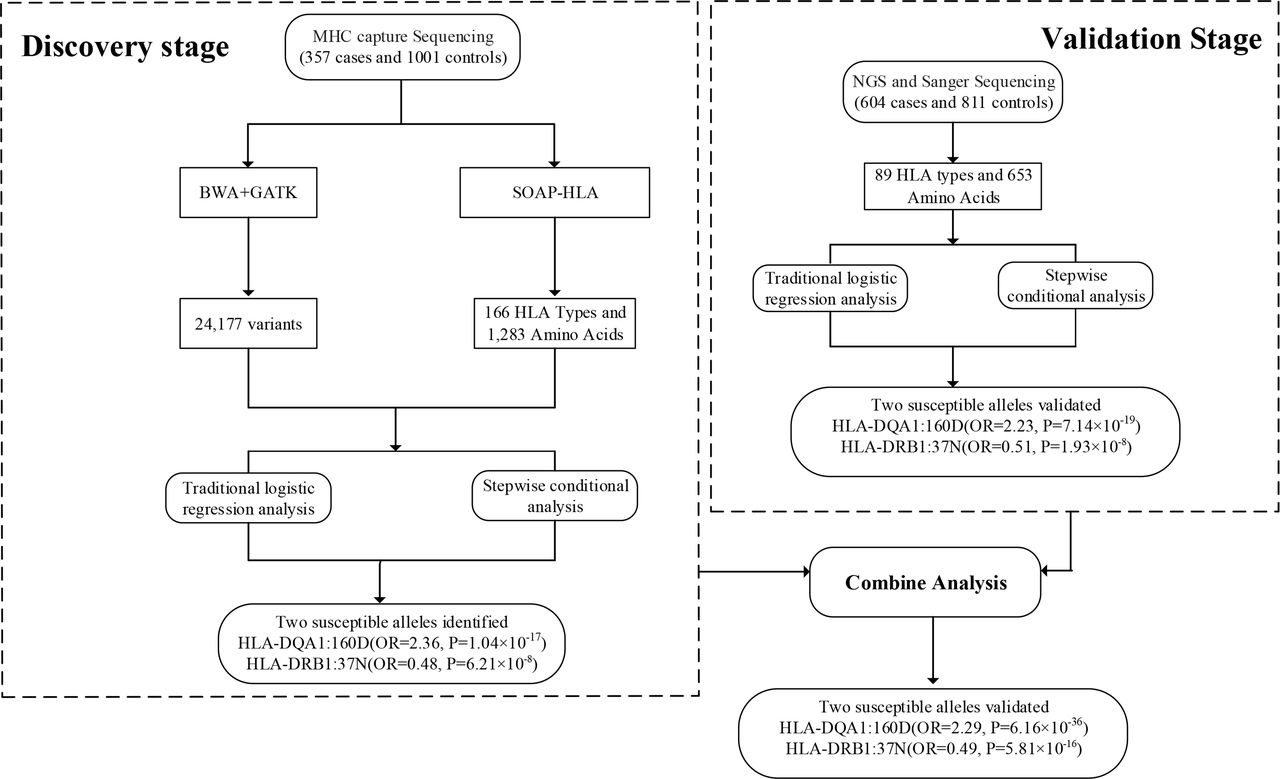
Two independent cohorts, including 1358 subjects for discovery cohort (357 cases and 1001 controls) and 1415 subjects for validation cohort (604 cases and 811 controls), were enrolled in the study. The schematic diagram of the study design is shown in figure 1. The baseline demographic characteristics of the patients and healthy controls are summarised in table 1.

Schematic diagram of the study design for the discovery and validation of the MHC region in ACPA-positive RA. ACPA, anticitrullinated protein antibodies; BWA-GATK, Burrows-Wheeler Aligner-Genome Analysis Toolkit; HLA, human leukocyte antigen; MHC, major histocompatibility complex; NGS, next generation sequencing; RA, rheumatoid arthritis; SOAP-HLA, Short Oligonucleotide Analysis Package-human leukocyte antigen.
Demographic characteristics of the study cohorts
All patients and healthy individuals were Han Chinese. Patients satisfied the American College of Rheumatology 1987 revised criteria for a diagnosis of RA,14 and were recruited from the Department of Rheumatology and Immunology at Peking University People’s Hospital. All cases were ACPA-positive patients with RA. Among cases, a total of 558 X-ray sets of hands were available. All X-rays were chronologically scored for assessment of bone destruction, as described previously.15 16
MHC target sequencing and variant calling
The MHC region was sequenced using the targeted sequencing methodology, as described previously.17 The samples were sequenced with standard 2×90 bp paired-end reads on the Illumina HiSeq 2000 sequencer. Sequencing data for the 1001 healthy controls were cited and selected by adjusting with age and sex from a recent publication of psoriasis project.18 Sequence reads were aligned to the National Center for Biotechnology Information (NCBI) human genome reference assembly (Build 37) using Burrows-Wheeler Aligner (V.0.5.9). On average, the MHC region was sequenced to a mean depth of 94 X, with 96.6% covered by at least one read and 93.1% covered by at least ten reads for cases, and a mean depth of 69 X for controls. Although the sequencing depth in cases and controls was differed, there was no bias for performance of variant calling and HLA typing according to previous studies.17 18 The variant calling and quality control of the target sequencing are detailed in online supplementary materials and methods.
Supplemental material
HLA typing
In the discovery cohort, a total of 26 highly polymorphic HLA genes were genotyped according to the Short Oligonucleotide Analysis Package (SOAP)-HLA. SOAP-HLA is a flow of sequencing data analysis pipeline to type any HLA genes using capture sequenced data based on IMGT/HLA database with a high accuracy.17 The amino acid sequence was determined according to the IMGT/HLA database (V.3.22.0).
In the validation cohort, the subjects were genotyped for HLA-A, HLA-B, HLA- DRB1, HLA-DPB1 and HLA-DQA1. HLA-A, H LA -B, HLA-DRB1 and HLA -DPB1 were genotyped using the next-generation sequencing method.19 HLA-DQA1 were genotyped by sequencing of both exons 2 and 3 using the Sanger sequencing method.20
Statistical analyses
Using the same data processing and analysis,18 21 the logistic regression model was applied to test the association between disease and variants, adjusting by gender. We define the HLA variants by including the biallelic SNPs, Indels, classical HLA alleles and individual HLA amino acid polymorphisms. For individual HLA allele and amino acid variants, the association was determined after stratifying the data using the relative predispositional effect method.22 To evaluate the overall significance of the multiallelic HLA amino acid polymorphisms for respective positions, the omnibus association test was conducted using the R statistics program (detailed in online supplementary M&M). To assess the independent effects for the candidates identified in logistic analysis, the stepwise conditional regression model was applied by additionally including the most significant loci as covariates, as described in online supplementary M&M.
In the discovery stage, a p value less than 1.0×10−6 was suggested as the significance threshold.23 24 In the validation and combined stages, a genome-wide statistical significance threshold of p value less than 5.0×10−8 was applied.
Comparative modelling
The comparative modelling was conducted using Modeller V.9.14.25 The construction for comparative modelling is detailed in online supplementary M&M. The crystal structure of HLA-DR1 (DRA-DRB1*0101) (PDB ID: 1AQD) was employed as the template for comparative modelling of DQA1*0303-DQB1*0401.26 The overall sequence identity and similarity between DQA1*0303-DQB1*0401 and HLA-DR1 are 61.7% and 76.6%, respectively. The crystal structure of HLA-DR3 (DRA-DRB1*03:01) (PDB ID: 1A6A) was employed as the template for the comparative modelling for DRA-DRB1*13:01 and DRA-DRB1*13:02.27 The sequence identities of HLA-DR3 to DRA-DRB1*13:01 and DRA1-DRB*13:02 were 98.1% and 97.8%, respectively.
Results
HLA-DQα1:160D is the strongest genetic risk for ACPA-positive RA, discovery by MHC sequencing
We first conducted a capture sequencing of the whole MHC region in 357 patients with ACPA-positive RA and 1001 previously sequenced healthy individuals. After quality control, a total of 24 177 variants, 166 HLA types and 1283 amino acids were obtained (online supplementary tables 1 and 2), in which 584 variants (including SNPs, HLA types and amino acids) showed significant associations by a cut-off of p value less than 1.0×10−6 (online supplementary table 3). We found that the top association signal was mapped to HLA-DQA1, with a peak at HLA-DQα1 amino acid position 160 (DQα1:160D, p=1.04×10−17, OR=2.36, 95% CI 1.94 to 2.87), followed by DQα1:160A (p=6.25×10−17, OR=2.27, 95% CI 1.87 to 2.75) (figure 2A and online supplementary table 3). HLA-DRB1*0405 allele also showed a strong association with ACPA-positive RA, but fell out of the top 10 risk variants (p=7.94×10−14, OR=3.45, 95% CI 2.50 to 4.78) (online supplementary table 3). These results indicated that by sequencing the entire MHC region, we discovered HLA-DQα1:160D may be the top genetic risk for ACPA-positive RA in the Han population, instead of the well-known HLA-DRB1 *0405.
Supplemental material

Plots of stepwise conditional analysis for ACPA-positive RA in the MHC region for the discovery cohort and the combined cohort. (A) Each diamond represents −log10(p) of the variants, including SNPs, Indels, classical HLA alleles and amino acid polymorphisms of HLA genes. The dotted horizontal line represents the suggestive significance threshold of p=1×10−6. The bottom panel indicates the physical positions of the HLA genes on chromosome 6 (NCBI Build 37). (B) The association for each locus used for conditioning (HLA-DQA1, HLA-DRB1) is shown in red in each panel. For each panel, the horizontal axis shows the position of amino acid for each HLA gene, and the vertical axis shows the negative log10-transformed p values for association. The dashed horizontal line corresponds to the significance threshold of p=5×10−8. ACPA, anticitrullinated protein antibodies; HLA: human leukocyte antigen; MHC, major histocompatibility complex; NCBI: National Center for Biotechnology Information; RA, rheumatoid arthritis; SNPs, single nucleotide polymorphisms.
HLA-DRβ1:37N has an independent protective effect on ACPA-positive RA
By stepwise conditional analysis on HLA-DQα1:160D, the second signal was mapped to a list of SNPs, with a peak at rs9275376 (chr6_32668633_G_T) (online supplementary table 4). The SNPs are in high LD (r2=0.82), and majority of the variants are located in the intergenic region between DQA2 and DQB1 according to the NCBI RefSeq database. An immediate significant association was observed for HLA-DRβ1:37N following these intergenic SNPs (figure 2A and online supplementary table 4). Of note, HLA-DRβ1:37N has an independent protective effect on ACPA-positive RA (p=6.21×10−8, OR=0.48, 95% CI 0.37 to 0.63). DRβ1:96 hours also showed a significant association (p=6.27×10−7, OR=1.59, 95% CI 1.33 to 1.91). Conditioning on DRβ1:37N, the intergenic SNPs and DRβ1:96 hours lost their association and no additional independent association(s) reached the suggestive statistical significance threshold of p<1.0×10−6 (data not shown).
Supplemental material
To assess whether DQα1:160D and DRβ1:37N were independent of each other, we performed the conditional analysis starting from DRβ1:37N. As shown in online supplementary figure 1, after conditioning on DRβ1:37N, DQα1:160D displayed an even stronger association (p=6.38×10−23, OR=3.55, 95% CI 2.76 to 4.56). This indicates that DQα1:160D also has an independent effect on ACPA-positive RA.
The validation study confirms the findings discovered by capture sequencing
To validate the findings discovered by capture sequencing, we genotyped HLA-A, HLA -B, HLA-DRB1, HLA-DQA1 and HLA-DPB1 in an independent case–control cohort, consisting of 604 cases and 811 healthy subjects. In line with the findings in the discovery stage, DQα1:160D showed consistent top association in the validation panel (p=7.14×10−19, OR=2.23, 95% CI 1.87 to 2.66) (online supplementary table 5). After conditioning to DQα1:160D, although DRβ1:96 hours became the second independent signal (p=2.80×10−10, OR=1.68, 95% CI 1.43 to 1.97), it was followed by DRβ1:37N (p=1.93×10−8, OR=0.51, 95% CI 0.40 to 0.65) (online supplementary table 6). When conditioning on DRβ1:96 hours, DRβ1:37N lost the association (p=5.96×10−3) and vice versa (p=1.18×10−4; data not shown) according to the genome-wide statistical significance threshold of p<5.0×10−8.
Supplemental material
Supplemental material
Joint analysis of discovery and replication panels provided compelling evidence that HLA-DQα1:160D conferred the highest risk on ACPA-positive RA (p=6.16×10−36, OR=2.29, 95% CI 2.01 to 2.60) (figure 2B, online supplementary table 7). After conditioning to DQα1:160D, DRβ1:96 hours and DRβ1:37N showed similar independent effects (DRβ1:96 hours: p=4.90×10−16, OR=1.64, 95% CI 1.45 to 1.84; DRβ1:37N: p=5.81×10−16, OR=0.49, 95% CI 0.41 to 0.58; figure 2B and online supplementary table 8). After conditioning on DRβ1:96 hours, the DRβ1:37N lost the association (p=4.85×10−6). Similarly, when conditioning on DRβ1:37N, DRβ1:96 hours also lost the association (p=2.26×10−5), and there was no additional independent association(s) that reached the genome-wide statistical significance of p<5.0×10−8 (data not shown). When conditioning on DQα1:160D, other DRβ1 variants also showed significant associations, including the putative RA-risk variant DRβ1:11V (p=2.06×10−13, OR=1.79, 95% CI 1.53 to 2.09) and DRβ1:11D (p=2.99×10−10, OR=0.49, 95% CI 0.39 to 0.61; online supplementary table 8). Notably, DRβ1:11D is in strong LD with DRβ1:37N (r2=0.62; online supplementary table 9) and also showed a protective effect.
Supplemental material
Supplemental material
Supplemental material
Next, we performed the omnibus test to assess the overall significance of the multiallelic amino acids for respective positions. As shown in online supplementary table 10, position 13 in DRβ1 showed the top association (p=4.03×10−45), followed by position 11 (p=9.85×10−45), which is consistent with the previous findings.6 28 However, if focusing on the individual amino acids or classical HLA alleles, DQα1:160D remained the top association (p=1.82×10−38), followed by DQA1*03:03 (p=1.05×10−35) and DQα1:160A (p=2.72×10−35). DRB1*04:05 and DRβ1:11V also showed a strong association, but out of the top 10 risks (p=7.75×10−30 and p=1.25×10−28, respectively).
Supplemental material
As HLA-DRB1 was well recognised as the strongest risk for ACPA-positive RA in previous studies, we then investigated RA association at HLA-DRB1 separately. By stepwise conditional analysis, we found if DQA1 is excluded, the DRB1*0405, the amino acid variants at DRβ1:11, 13, 57, 74 and 71 could be strong risks for ACPA-positive RA (detailed in online supplementary results, supplementary figure 2 and supplementary table 11). We also compared the individual amino acid frequencies between Han Chinese and European populations. As shown in figure 3A, in the present study both DQα1:160D and DQα1:160A are common amino acids in Han Chinese and were significantly increased in cases. However, the two amino acids have not been detected in European population.6 For amino acid positions 11, 13, 57, 71 and 74 in DRβ1, the amino acid frequencies are similar between the two ethnic groups or case and controls, except for a few amino acids (figure 3B,C,D,E,F).
Supplemental material

Comparison of individual amino acid frequencies within DQα1:160 and DRβ1:11, 13, 57, 71 and 74 in Han Chinese and European populations. The individual amino acid frequencies are plotted in healthy controls (blue) and cases (red). The upper panel shows the amino acid frequencies in Han Chinese population (the data derived from the present study). The lower panel shows the amino acid frequencies in European population (the data cited from Raychaudhur et al‘s study6). (A) DQa1:160D and DQa1:160A are common amino acids in HanChinese and were significantly increased in patients with RA, but the two variantshave not been detected in European population. Similar frequencies of amino acids were observed at DRß1 position 11(B),13(C), 57(D), 71(E), and 74(F) between Han Chinese and European populations.
We next examined whether DQα1:160D and its encoding alleles, that is, DQA1*0302 and DQA1 *0303, confer a risk for the severity of joint damage in ACPA-positive RA. As shown in online supplementary figure 3, in the early disease stage, DQA1*0303 displayed high impact on radiographic scores in the smoking group (p=3.02×10−5; online supplementary figure 3A and detailed in online supplementary results).
Additional negative charge of D160α enhances the interaction of DQα1-DQβ1, leading to an increased T cell activation
MHC class II molecules could present in the form of either heterodimers or the dimer of the heterodimers (dimer of dimers).26 29 30 The dimer of dimers appears to play an important role in T cell response to low affinity antigens by enhancing overall affinity between MHC/peptide and T cell receptor (TCR).30 31 The electrostatic interactions between the interface residues of the dimer of dimers are critical for maintaining structural stability as well as T cell activation.32 According to sequence analysis, D160α locates away from antigen binding groove and should impose little influence on epitope binding. To investigate the potential function of DQα1:160D, we constructed the dimer of dimer structure for DQA1*0303-DQB1*0401 by comparative modelling, as it is the major D160α encoding haplotype in Han Chinese (69.0% from our data and 56% in the Han MHC database).18 As shown in figure 4A, D160α is adjacent to the dimer of dimer interface. Strong electrostatic interactions are observed between negative DQα1 interface residues (D161α, E182α and E184α) and positive DQβ1’ interface residues (R105β, H111β and H112β). Compared with non-charged A160α or S160α coded by other DQA1 alleles, additional negative charge of D160α further enhances the interaction between DQα1 and DQβ1’, which may lead to an increased T cell activation (figure 4A).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Dimer of dimer structure of DQA1*0303-DQB1*0401 (A) and the electrostatic potential surface and the pocket structure of DRA1-DRB1*03:01, DQA1*0303-DQB1*0401 and DRA1-DRB1*13:02 (B). (A) Overall dimer of dimer structure of DQA1*0303–DQB1*0401(left panel): one dimer is composed of DQα1 (green) and DQβ1 (cyan), whereas the other dimer is composed of DQα1’ (purple) and DQβ1’ (yellow). The interface of the dimer of dimer structure (right panel): DQα1 and DQβ1’ residues involved in the interaction are displayed as sticks and their carbon atoms are coloured in green and yellow, respectively, whereas nitrogen and oxygen atoms are coloured in blue and red, respectively. (B) The electrostatic potential surface of DRA1-DRB1*03:01, DRA1-DRB1*13:01 and DRA1-DRB1*13:02 (upper panel). All structure models are displayed by surface. The positive, neutral and negative regions are coloured in blue, white and red, respectively. The P9 pocket structure of DRA1-DRB1*03:01, DRA1-DRB1*13:01 and DRA1-DRB1*13:02 (lower panel). HLA molecules and pocket residues are shown as grey cartoon and green sticks, respectively. HLA, human leukocyte antigen.
The negatively charged P9 pockets from DRβ1:37N encoding alleles benefit electrostatic interaction and epitope P9 arginine binding
It is well accepted that HLA-DRB1 susceptibility alleles are strongly associated with ACPA-positive RA, and its encoded molecules preferentially present the citrullinated autoantigens.33 The susceptibility is determined by the electrostatic property of antigen binding pockets and the ability to differentially recognise citrullinated antigens. The positively charged antigen binding pocket in RA-susceptible alleles preferentially accommodate citrullinated antigens, whereas electronegative or electroneutral pockets in RA-resistant alleles can bind both arginine and citrullined antigens.34 The amino acid residues at position 37 in DRβ1 are located within the P9 pocket of antigen binding groove. By sequence analysis, we found four alleles containing an asparagine at position DRβ1:37 (Asn37β or 37N), including DRB1*03:01, DRB1 *09:01, DRB1 *13:01 and DRB1 *13:02. Although DRB1*0901 was reported to be the risk allele for RA,10 it was associated with a low level of anti-cyclic citrullinated peptide (CCP) antibodies in patients with RA or anti-CCP-negative RA.35 36 We then constructed the crystal structure for other allele-containing haplotypes by comparative modelling. As shown in the bottom panel of figure 4B, the DRA1-DRB1*03:01, DRA1-DRB1*13:01 and DRA1-DRB1*13:02 share identical P9 pocket, which is composed of Asn69α, Met73α, Tyr30β, Asn37β, Val38β and Asp57β. The electrostatic potential surface analysis as shown in the upper panel of figure 4B indicates that P9 pockets of the three haplotypes are negatively charged and could benefit electrostatic interaction with epitope P9 arginine. Collectively, all three alleles containing negatively charged P9 pocket of Asn37 could bind the epitope P9 arginine and thus may result in a decreased RA susceptibility.
As DRβ1:96 hours and DRβ137N have similar effects and cannot be distinguished by statistical analysis, we then sought to predict the potential function for DRβ1:96 hours and found the crystal structure of HLA-DR11 was available (DRA-DRB1*1101, one of major encoding alleles for DRβ1:96 hours in Han Chinese). As shown in online supplementary figure 4, unlike DRβ1:37N, DRβ1:96 hours (PDB code: 6CPL) locates far away from either antigen binding groove or the dimer of dimer interface. It may indicate that DRβ1:96 hours has little impact on either epitope binding or the structural stability of the dimer of dimers.
Discussion
A key finding of this study is the major genetic influence of HLA-DQα1:160D, instead of the well-described HLA-DRB1 alleles, on ACPA-positive RA in Han ancestry. HLA-DRβ1:37N is an independent protective factor for ACPA-positive RA. The novel findings are confirmed in an independent case–control cohort by classical HLA genotyping.
Previously, a number of studies have reported that the major genetic risk for ACPA-positive RA came from certain HLA-DRB1 alleles.37–39 However, other HLA genes have also been linked to ACPA-positive RA.6 11 Thus, it is difficult to draw definitive conclusion concerning different HLA genes in susceptibility to ACPA-positive RA. Furthermore, several examples of RA genetic heterogeneity have been shown between Asian and Caucasian populations. One of evidence is that the DRB1*0401 and DRB1 *0405 are considered as the top RA-risk alleles in Caucasians and East Asians, respectively.5 8 10 The non-HLA gene PTPN22 has been confirmed to be a common genetic risk for multiple autoimmune diseases, including RA.40 The disease-associated SNP in PTPN22 (rs2476601 C>T) results in a substitution at amino acid 620 (Arg620Trp), consequently leading to a reduced TCR signalling.41 Interestingly, the T allele is almost absent in African–American and Asian populations,42 a clear evidence of genetic heterogeneity between Europeans and Asians. PADI4 is another good example of genetic heterogeneity in RA susceptibility. PADI4 is one of several peptidylarginine deiminase enzymes that catalyse the conversion of arginine residues into citrulline, and thus may be linked to production of anticitrulline antibodies. The association between the PADI4 polymorphism and ACPA-positive RA has been reported in several Asian populations.43 44 In contrast, conflicting results were seen in European populations.45 46
In the present study, we identified HLA-DQα1:160D as the strongest and independent genetic risk for ACPA-positive RA in Han population. Our novel finding could be rationally explained by the fact that DQα1:160D is encoded by two alleles, that is, DQA1*0302 and DQA1 *0303. Although there were several large-scale RA–MHC association studies that included the DQA*03 alleles in the reference panels (European ancestry reference panel and Pan-Asian reference panel, respectively),6 7 11 the DQA1*03 alleles were only presented in either two-digit resolution (DQA1*03) and/or four-digit resolution of DQA1*03:01. The possible explanation for the absence of the DQA1*03:02 and DQA1 *03:03 could be that the reference panels of previous large-scale RA-MHC studies were constructed by using the SNP2HLA, which inputs SNPs with allele frequencies ≥1% and removes the markers (eg, HLA alleles and amino acids) with rare allele frequencies (<0.01%).47 Although there was a report showing the frequencies of DQA1*03:02 and DQA1 *03:03 were 0.017 and 0.082 in UK Europeans, the study had a very small sample size (n=254) and published after the SNP2HLA study.48 Thus, we speculate that the SNPs tagging DQA1*03:02 and DQA1 *03:03 may not be included in the SNP2HLA imputation due to the low frequencies (minor allele frequency, MAF <1%). These could help to explain why DQα1:160D and its coding alleles *0302 and *0303 have not been detected or suggested to be risk factors for RA in previous studies, including a recent large-scale HLA imputation study of ACPA-positive RA in Japanese population.28
Since HLA-DRB1 was well recognised as the top risk for ACPA-positive RA in previous studies, we then tested the association at HLA-DRB1 separately. We showed that if DQA1 was not included in analysis, the allele DRB1*0405, amino acid variants at DRβ1:11, 13, 57, 74 and 71 could be strong risks for ACPA-positive RA, which is consistent with the prevous findings.7 Furthermore, smoking has been reported as a risk factor linked to RA susceptibility and severity. This risk was increased due to the gene–environment interaction between smoking and DRB1 alleles. Consistent with previous findings,9 we also found the smoking DRB1*0405 carriers had increased radiographic damage in ACPA-positive patients with RA at an early stage of disease. We further showed that one of DQα1:160 coding alleles DQA1*0303 has high impact on radiographic severity, especially in smoking patients and in early disease. Our data thus support the notion that smoking, in the presence of RA-risk genetic background, may trigger immunity to citrullinated proteins and lead to RA development and accelerate joint damage.
In the present study, we mainly focused on SNPs, classical HLA alleles and the individual amino acid variants rather than amino acid positions, since a particular amino acid(s) may have potential biological function(s). Furthermore, different amino acids at the same position may insert different functions. For example, the residues in DRB1*04:01 (Val11β and His13β) and DRB1*01:01 (Leu11β and Phe13β) would prevent the accommodation of arginine (Arg) at the P4 pocket.49 In contrast, the P4-Arg is stabilised by H-bonds with Ser11β, Ser13β and Tyr30β.50 In Raychaudhuri et al’s6 study, they found the five individual amino acids in three HLA proteins could explain most of the association between MHC and seropositive RA in Europeans. In the present study, we observed that, although DQα1:160 locates away from antigen binding groove and may have little influence on epitope binding, it is adjacent to the DQα1-DQβ1 dimer of dimer interface. A previous study has reported the amino acid substitutions in the dimer of dimer interface of HLA-DRβ1 inhibited CD4+ T cell activation.51 Here we showed that the additional negative charge introduced by D160α further enhances the interaction between DQα1 and DQβ1 and stabilises the dimer of dimers, which may lead to an increased T cell activation.
The DRβ1:37 residues are located within pocket P9 on the beta-sheet floor with their side chains oriented into the peptide-binding groove. Modification of DRβ1:37 residues is sufficient to alter the T cell receptor peptide recognition.52 53 Pocket P9 of HLA-DRβ has been linked to autoimmune diseases, such as primary Sjogren’s syndrome and primary sclerosing cholangitis.54 55 In the present work, by electrostatic potential surface analysis, we showed that three DRβ1:37N encoding alleles, *03:01, *13:01 and *13:02, have negatively charged P9 pocket, which benefits electrostatic interaction and could bind with epitope P9 arginine. Previously, Kampstra et al 56 also reported that DRB1*03:01 had no preference for citrulline in any of the pockets, while it preferred arginine residues as anchors in pockets 6 and 9. Thus, it could at least partly explain the protective effect of DRβ1:37N for ACPA-positive RA. In contrast, DRβ1:96 hours locates far away from either the antigen binding groove or the dimer of dimer interface. It may indicate that DRβ1:96 hours has little impact on antigen presentation or structural stability of the dimer of dimers.
In summary, by sequencing of the entire MHC region for discovery and HLA genotyping for validation in two independent cohorts, our study demonstrates that HLA-DQα1:160D, instead of HLA-DRB1*0405, confers the greatest independent genetic risk for ACPA-positive RA in Chinese Han. Our study also illustrates the value of deep sequencing for fine-mapping disease risk variants in MHC region. However, one should bear in mind that current findings are mainly built on statistical significance and the functional prediction. Further functional studies are needed to fully understand its contribution to RA susceptibility.
Acknowledgments
We thank the staff from the Department of Rheumatology and Immunology, People’s Hospital, Peking University, for recruiting patients and healthy controls, the staff from the Computing Platform of the Center for Life Sciences, Peking University, for assisting the functional prediction analysis, and the staff from BGI-Shenzhen who contributed to the technical assistance. We wish to thank the patients and healthy volunteers for their cooperation and for giving consent to participate in this study.
References
Footnotes
JG, TZ, HC, XL and HL contributed equally.
Handling editor Josef S Smolen
Contributors JG, XX and ZL conceptualised and designed the study. XJZ and LK participated in the study design and supervised manuscript writing. JG, TZ and HC coordinated and supervised the study teams. JG, XWL, TZ, HL and HC conducted data management and manuscript preparation. TZ, XWL, YWZ, XM and HJY conducted the statistical analyses. HMY, HJJ, JW, LS, LP, LHL, LL and KY participated in data interpretation and manuscript writing. ML, YDZ, YW, XS, FH, YD, MZ, HJ, XuL, YH, XiL, YY, XW, XZ and YS conducted sample selection and participated in data management. All coauthors edited and reviewed the final version of the manuscript.
Funding This work was supported in part by the National Key Basic Research Program of China (973 Program) (No 2014CB541901), the National Natural Science Foundation of China (No 81120108020, No 31711530023, No 31670915, No 31870913, No 31470875, No 31270914, No 31530020, No 31700794, No 81401329, No 81771678, No 81471601, No 81671604), the National Key Research and Development Program of China (No 2016YFA05022300), Beijing Natural Science Foundation (No 7162192), and Shenzhen Municipal of Government of China (No CXB201108250094A).
Competing interests None declared.
Patient consent for publication Obtained.
Ethics approval The study was approved by the Medical Ethics Committee of Peking University People’s Hospital.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as online supplementary information.