Article Text

Abstract
Objectives Cyclophosphamide induction regimens are effective for antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV), but are associated with infections, malignancies and infertility. Mycophenolate mofetil (MMF) has shown high remission rates in small studies of AAV.
Methods We conducted a randomised controlled trial to investigate whether MMF was non-inferior to cyclophosphamide for remission induction in AAV. 140 newly diagnosed patients were randomly assigned to MMF or pulsed cyclophosphamide. All patients received the same oral glucocorticoid regimen and were switched to azathioprine following remission. The primary endpoint was remission by 6 months requiring compliance with the tapering glucocorticoid regimen. Patients with an eGFR <15 mL/min were excluded from the study.
Results At baseline, ANCA subtype, disease activity and organ involvement were similar between groups. Non-inferiority was demonstrated for the primary remission endpoint, which occurred in 47 patients (67%) in the MMF group and 43 patients (61%) in the cyclophosphamide group (risk difference 5.7%, 90% CI −7.5% to 19%). Following remission, more relapses occurred in the MMF group (23 patients, 33%) compared with the cyclophosphamide group (13 patients, 19%) (incidence rate ratio 1.97, 95% CI 0.96 to 4.23, p=0.049). In MPO-ANCA patients, relapses occurred in 12% of the cyclophosphamide group and 15% of the MMF group. In PR3-ANCA patients, relapses occurred in 24% of the cyclophosphamide group and 48% of the MMF group. Serious infections were similar between groups (26% MMF group, 17% cyclophosphamide group) (OR 1.67, 95% CI 0.68 to 4.19, p=0.3).
Conclusion MMF was non-inferior to cyclophosphamide for remission induction in AAV, but resulted in higher relapse rate.
Trial registration number NCT00414128.
- ANCA-associated vasculitis
- induction therapy
- cyclophosphamide
- mycophenolate
- randomised trial
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Cyclophosphamide remains the first-line induction remission treatment for antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis for many patients but is linked with infertility, infection and malignancy.
Mycophenolate mofetil (MMF) has been shown in small studies to have high remission rates.
What does this study add?
This study is the largest to show with sufficient power that remission rates with MMF are non-inferior to pulsed cyclophosphamide but this may be associated with a higher rate of relapse.
How might this impact on clinical practice or future developments?
MMF induction therapy in patients at low risk of relapse, such as those with myeloperoxidase-ANCA, may be a suitable alternative to cyclophosphamide.
Background
Antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV),1 which includes granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA), is a rare potentially life-threatening multisystem autoimmune disease. They are frequently grouped together for the purpose of treatment trials given their similar initial responses to standard therapy.2 3 Treatment for AAV comprises remission induction and maintenance regimens.2 The European League Against Rheumatism (EULAR) guidelines for the treatment of AAV suggest the use of cyclophosphamide (CYC) or rituximab for remission induction therapy in new-onset organ-threatening or life-threatening AAV in combination with glucocorticoids.4 CYC with high-dose glucocorticoids has been the standard remission induction therapy for severe AAV for over 30 years with remission rates of 80%–90%5 6 and a current 1-year mortality of 10%–25%.7 However, CYC is toxic causing infertility and malignancy. Rituximab is associated with remission induction rates similar to those achieved with CYC and similar relapse rates over 18–24 months of follow-up.8–11 However, the biological effect of rituximab is long and variable, and rituximab has been associated with hypogammaglobulinaemia in AAV.12 Due to its high cost the use of rituximab is restricted in some countries.13 14 For non-organ-threatening AAV, EULAR recommends methotrexate or mycophenolate mofetil (MMF) in combination with glucocorticoids, although the level of evidence is rated as 1B, requiring further studies.4 Methotrexate has efficacy similar to CYC for remission induction in non-severe AAV, but its toxicity precludes use in renal impairment.15 16 MMF is an alternative oral immunosuppressant with lymphocyte selective suppressive effects with a short duration of action, can be used in renal disease and unlike CYC is not associated with urothelial malignancy or infertility. Small studies have suggested that MMF has efficacy for remission induction in AAV, particularly in myeloperoxidase (MPO)-ANCA disease.17 18 Understanding the role of MMF as a remission induction agent in AAV remains important. We conducted a randomised trial of adult and paediatric patients to investigate whether MMF was non-inferior to CYC for remission induction in new patients with AAV.
Methods
Study design and patients
This trial was an open-label, two-group, parallel-design, randomised, non-inferiority trial involving 132 adult patients from 21 sites in six countries in Europe, Australia and New Zealand, and eight paediatric patients from four sites in the UK. All patients/parents provided written informed consent; and written assent where appropriate. Inclusion in this study required a new diagnosis of active AAV (GPA or MPA)1 with either a positive ANCA or histologically proven disease (see protocol for full inclusion details). Patients were excluded if they were aged <6 years, had imminently life-threatening vasculitis, rapidly declining renal function or an estimated glomerular filtration rate (eGFR) <15 mL/min/m2, or had received >2 weeks of oral CYC or MMF or more than 1 pulse of intravenous CYC (15 mg/kg). The trial protocol is available at http://vasculitis.org/images/documents/mycyc.pdf.
Patients were allocated in a 1:1 ratio to MMF or CYC using a concealed system of minimisation by: age >60 years, the planned use of additional therapy with plasma exchange or solumedrol >0.5 g at randomisation, eGFR <30 mL/min/m2 or ≥30 mL/min/m2 with an allocation probability of 0.8. Although the minimisation procedure did not include site as a stratification factor, the degree of balance of treatments within the sites was reasonable (online supplementary table 1).
Supplemental material
The trial was sponsored by Cambridge University Hospitals NHS Foundation Trust. Vifor Pharma (previously Aspreva Pharmaceuticals) provided a research grant to cover the trial and MMF costs. The trial protocol was designed by the ‘MYCYC’ trial steering committee, and received ethical and regulatory approval in each participating country. The trial was conducted according to the European Union (EU) clinical trials directive (Directive 2001 EU/20/EC) (EUDRACT 2006-001663-33). Trial data are stored by the trial management committee at Addenbrooke’s Hospital, UK.
Treatments
After randomisation, both groups received the same oral tapering glucocorticoid regimen (prednisolone 1 mg/kg/day initially, reducing to 5 mg/day at the end of 6 months (online supplementary figure 1D)). Adult patients in the MMF group received MMF 2 g/day, with dose increases to 3 g/day permitted for uncontrolled disease at 4 weeks. Patients aged less than 17 years received a body surface area-based MMF dosing regimen. Patients in the CYC group received intravenous pulsed CYC as given in the CYCLOPS trial (15 mg/kg every 2–3 weeks with reductions for age and renal function).6 9 All patients were switched from their assigned study treatment to oral azathioprine (AZA) 2 mg/kg/day after remission had been achieved, between 3 and 6 months. AZA with prednisolone 5 mg/day was continued until study end at 18 months.
Outcomes
The primary outcome was remission by 6 months. Remission was defined as the absence of disease activity with a Birmingham Vasculitis Activity Score (BVAS) 2003 of zero on two consecutive occasions at least 1 month apart and adherence to the prednisolone taper. Secondary efficacy endpoints were time to remission, remission by 6 months irrespective of glucocorticoid adherence, progressive disease, relapse, cumulative glucocorticoid dosing, change in eGFR, Vasculitis Damage Index (VDI)19 and ANCA positivity at 6 months. Planned subgroup analyses were the effect of eGFR, age and additional intravenous methylprednisolone and/or plasma exchange prerandomisation on remission, and ANCA subtypes, on remission and relapse. Safety outcomes were serious adverse events, serious infections, end-stage renal disease (ESRD), death, malignancy, cardiovascular, thromboembolic and serious disease-related events. Outcomes were adjudicated by a committee blinded to study group assignment.
Assessments
Assessments were performed at 0, 1, 1.5, 3, 4.5, 6, 9, 12 and 18 months and at the time of a relapse. Relapses could only occur after an initial remission (absence of disease activity, irrespective of glucocorticoid compliance, at any time during trial follow-up). Patients who did not achieve an initial remission were excluded from relapse analyses. Relapses were defined as the recurrence or new appearance of any disease activity, as reflected by a BVAS 2003 >0. Major relapse required the presence of one or more major BVAS items. Renal function was assessed using eGFR, calculated using the four-variable Modified Diet in Renal Disease equation in adults20 or Haycock-Schwartz formula in patients aged <16 years.21 ESRD was defined as dialysis dependence for 6 weeks or more without subsequent recovery of renal function. Progressive disease was defined as ongoing disease activity of sufficient severity to necessitate therapy escalation with a change in immunosuppression or intravenous methylprednisolone before remission. Serious adverse events were collected as defined by the European Medicines Agency and Food and Drug Administration. ANCA negativity was determined by the reference range of the local laboratory for both indirect immunofluorescence and ELISA.
Statistical analysis
The sample size estimate was based on a non-inferiority design. We assumed a remission rate of 85% with CYC and specified a 12% absolute risk difference (RD) as the non-inferiority margin (ie, remission rate <73%) for MMF. Using these assumptions, we calculated that 124 patients were required to meet non-inferiority for the primary remission endpoint with a power of 80%, and a significance level of 5% in a non-inferiority test.22 Allowing for a 10% dropout rate we recruited 140 patients.
All endpoint analyses were by intention to treat with an additional prespecified per protocol analysis of the primary endpoint. The primary and secondary remission endpoints (non-inferiority) were assessed by calculating the RD of remission with corresponding two-sided 90% CIs, consistent with the Consolidated Standards of Reporting Trials extension for reporting of non-inferiority trials.23 For the primary analyses, no attempts were made to impute missing data. Data were censored at withdrawal, loss to follow-up or death. Time to event analyses of remission (non-inferiority) were performed using a Cox proportional hazards model with an HR of 0.85 as the non-inferiority margin. Relapse rates (superiority) were compared by calculating the incidence rate ratio (IRR; relapses per patient per year) and corresponding 95% CI with significance estimates derived from the binomial distribution test. For safety and other efficacy endpoints comparison of proportions was performed using the Fisher mid-p test, as recommended.24 All continuous variables are presented as mean±SD or median (IQR) as appropriate to their distribution, and categorical variables are presented as count (%). All analyses were conducted using Stata SE V.15 (College Station, TX).
Results
Patients
Between March 2007 and July 2011, a total of 140 patients were enrolled in the study (66 adults and four children in each treatment group). The four children recruited to the CYC group were aged 14, 16, 14 and 15 years and the four recruited to the MMF group were 10,16, 12 and 13 years old. All patients received their allocated treatment and were retained for the primary analysis. By the end of the 6-month treatment period, four in each group had died, and three in the MMF group and two in the CYC group had been lost to follow-up or had withdrawn consent (figure 1, table 1). Fifty-eight patients received at least 6 pulses of CYC, of whom 23 had 7–10 pulses. CYC was terminated early in six (two infection, two intolerance, one ESRD, one no reason), and six died or withdrew from the trial prior to 3 months. The maximum dose of MMF received by patients was 2 g in 76%, 6% received >2 g and 18% received <2 g. MMF was withdrawn due to intolerance in four patients due to incontinence, rash, diarrhoea and reason not specified. At 18 months, 52 patients, 26 from each study group, were not receiving AZA. This was due to drug intolerance in 11 patients in the CYC group and 15 patients in the MMF group.
Baseline characteristics of the patients at trial entry
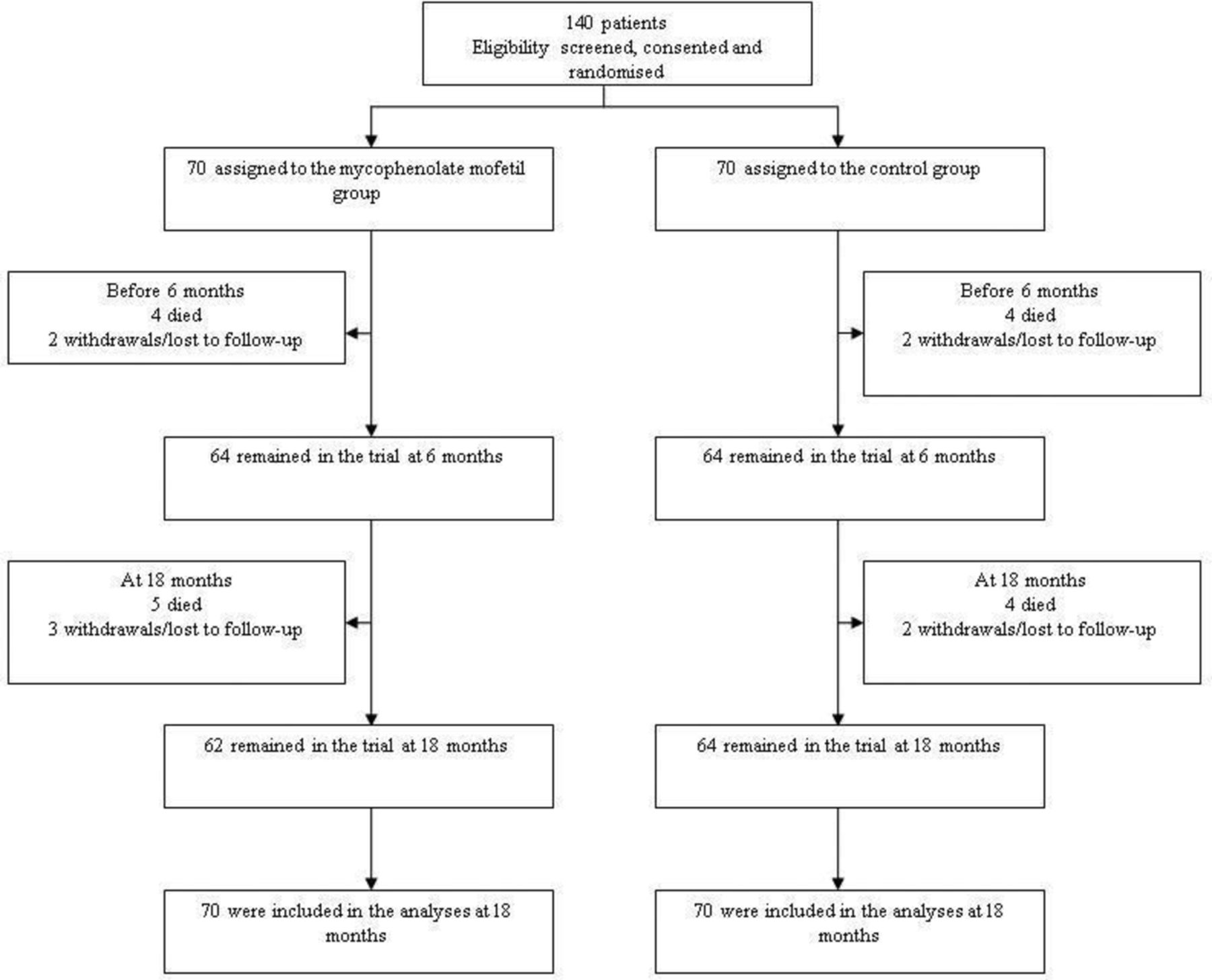
Randomisation and inclusion in the analysis at 18 months.
Primary outcome
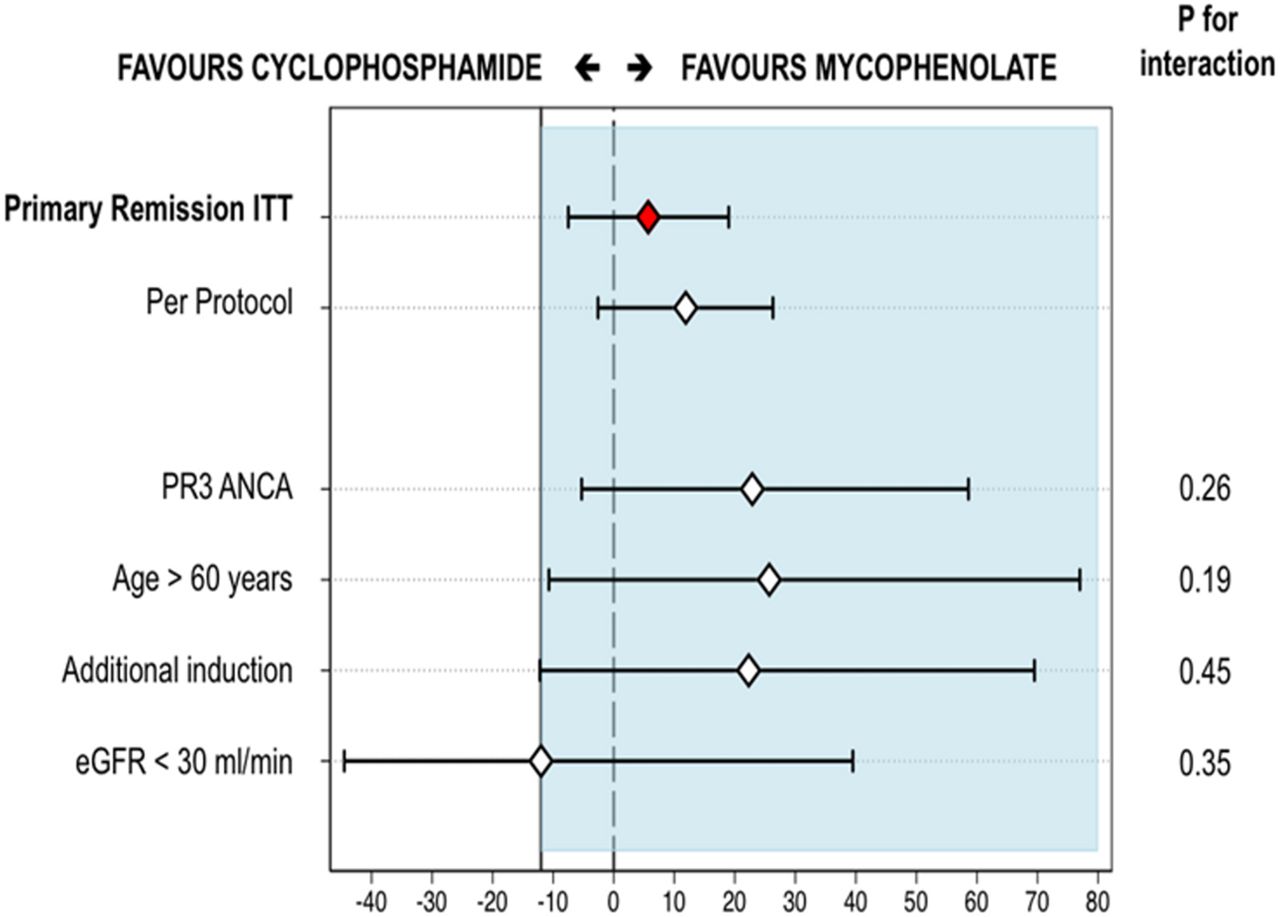
The primary endpoint of remission with glucocorticoid compliance within 6 months occurred in 47 (67%) patients, including one child, in the MMF group, and 43 (61%), including one child, in the CYC group (RD 5.7%, 90% CI −7.5% to 19%). Given the specified non-inferiority margin of −12%, the lower bound of the 90% CI of −7.5% established non-inferiority (figure 2).

Absolute risk ratio for the primary remission endpoint, per-protocol and subgroup analyses. The lower bound 90% CI did not cross the non-inferiority margin of 12% for the primary endpoint and per-protocol analyses demonstrating non-inferiority. The lower bound 90% CI only crossed the non-inferiority margin for patients with eGFR <30 mL/min but the upper bound 90% CI exceeded 0. This would be described as ‘inconclusive’ and given this is a secondary analysis of a subgroup we are unable to draw any inference from this other than the p value for interaction being non-significant. The diamonds represent the absolute risk ratio, horizontal black lines represent 90% CIs. The left side of blue shaded area represents the lower limit of non-inferiority margin (−12%). ANCA, antineutrophil cytoplasmic antibody; eGFR, estimated glomerular filtration rate; ITT, intention to treat.
In a prespecified analysis restricted to per-protocol treated patients, 43 remissions (74%) occurred in 58 mycophenolate patients, compared with 33 remissions (62%) in 53 CYC patients (RD 11.9%, 90% CI −2.6% to 26.3%, non-inferior) (figure 2). There was no evidence of interaction by PR3-ANCA positivity, age, renal function and the use of additional induction therapies with the primary endpoint (figure 2).
Secondary efficacy outcomes
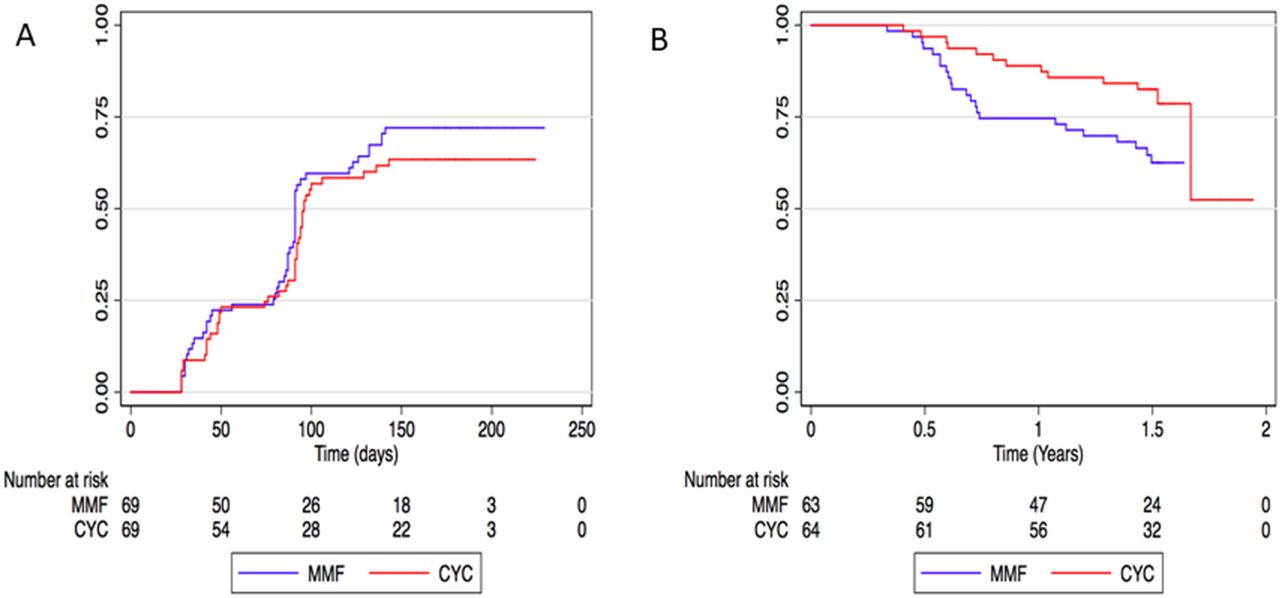
Secondary efficacy outcomes are summarised in figure 3 (online supplementary table 2 and figure 1). The time to primary remission in the MMF group (median 91 days, IQR 44–95) was non-inferior to the CYC group (median 87 days, IQR 42–91), since the lower bound of the 90% CI did not cross 0.85 (HR 1.27 (90% CI 0.89 to 1.79)).
Supplemental material

{kind=link}
{kind=link}
![[annrheumdis-2018-214245-supp2.JPG]](https://ard.bmj.com/content/annrheumdis/78/3/399/DC2/embed/inline-supplementary-material-2.jpg?download=true){kind=link}
{kind=link}
Remission and relapse. (A) Time to primary remission. Primary remission was remission with no disease activity and glucocorticoid protocol compliance. Analysis was censored at the first of the following events: remission (first Birmingham Vasculitis Activity Score (BVAS) of zero), 6-month study visit, withdrawal or death. (B) Time to first relapse. Relapse could only occur after an initial remission. Remissions for this analysis are not restricted to the first 6 months of follow-up, but represent remissions occurring at any time point after randomisation irrespective of glucocorticoid compliance. Time to first relapse was significantly shorter in the mycophenolate mofetil group. CYC, cyclophosphamide; MMF, mycophenolate mofetil.
Remission irrespective of steroid compliance within 6 months occurred in 61 patients (87%) in the MMF group and 55 (79%) in the CYC group (RD 8.6%, 90% CI −1.8% to 19%). Remission at any time during trial follow-up irrespective of steroid compliance occurred in 63 patients (90%), including two children, in the MMF group, and 64 (92%), including two children, in the CYC group (RD −1.4%, 90% CI −9.5% to 6.6%).
There were more relapses after remission in the mycophenolate group (23/63 patients; 4 major and 19 minor relapses) compared with the CYC group (13/64 patients; 3 major and 10 minor relapses, IRR 1.97, 95% CI 0.96 to 4.23, p=0.049). Relapse-free survival was shorter in the mycophenolate group (HR 2.14, 95% CI 1.07 to 4.31, p=0.03). A post hoc subgroup analysis found the higher relapse rate in MMF patients was accounted for by more relapses in PR3-ANCA patients, but not MPO-ANCA patients (online supplementary Figure 2). There was no evidence that the effect of MMF on relapse differed by ANCA subtype (p=0.52 for interaction).
Supplemental material
![[annrheumdis-2018-214245-supp4.JPG]](https://ard.bmj.com/content/annrheumdis/78/3/399/DC3/embed/inline-supplementary-material-3.jpg?download=true){kind=link}
Remission irrespective of steroid compliance within 6 months occurred in 61 patients (87%) in the MMF group and 55 (79%) in the CYC group (RD 8.6%, 90% CI −1.8% to 19%). Remission at any time during trial follow-up irrespective of steroid compliance occurred in 63 patients (90%) in the MMF group and 64 (92%) in the CYC group (RD −1.4%, 90% CI −9.5% to 6.6%).
Progressive disease necessitating rescue therapy before remission occurred in 5 patients (7%) in the MMF group and 8 (11%) in the CYC group (p=0.56). At 6 months, 26 of 65 (40%) patients in the MMF group were ANCA negative, and 21 of 65 (32%) patients in the CYC group were ANCA negative (risk ratio 1.23, 95% CI 0.78 to 1.96, p=0.36).
There was no statistically significant difference in cumulative glucocorticoid exposure during the trial (MMF 6194±317 mg, CYC 5800±234 mg, p=0.32) (online supplementary figure 1A). Two patients in both groups progressed to ESRD and eGFR at 18 months did not differ between groups (MMF group 68±4 mL/min, CYC group 64±4 mL/min, p=0.46) (online supplementary figure 1B). There was no difference in disease and treatment-related damage assessed by the VDI at study end between the two groups (MMF=1, IQR 1–3; CYC=2, IQR 1–3; p=0.80).
Safety outcomes
Serious adverse events occurred in 35 in the MMF group (50% patients, 73 events) and 28 in the CYC group (40% patients, 64 events) and are summarised in table 2. There were no significant differences in serious infections, death, thromboembolism, malignancy or serious disease-related events between the two groups.
Serious adverse events
Five mycophenolate patients died (7%) (causes of death were cardiac n=1, infections n=2 and other n=2) and four CYC patients died (6%) (causes of death were cardiac n=1, infections n=2 and other n=1) (OR 1.27, 95% CI 0.26 to 6.68, p=1.0). Median age at death was 75 years (range 73–82 years) in the MMF group and 83 years (range 63–85 years) in the CYC group. Malignancies were liver metastases of unknown primary in a 74 year-old in the mycophenolate group and a malignant melanoma in a 63 year-old in the CYC group.
Discussion
In this randomised trial of remission induction in AVV, excluding patients on dialysis or with life-threatening disease, MMF was non-inferior to pulsed CYC. The relatively low remission rate for the primary outcome can be attributed to the stringent requirement for adherence to glucocorticoid taper as shown by others,8 and the higher rate of the secondary endpoint of remission irrespective of glucocorticoid adherence is consistent with previous reports where the glucocorticoid taper was not a component of the remission definition.6 25 Our results demonstrate that MMF represents an alternative to CYC for remission induction in AAV. This study provides further evidence to support the EULAR guidelines on management of AAV.
Our findings of the efficacy of MMF for remission induction are consistent with previous MMF induction studies in AAV.18 26 27 After remission, relapses occurred earlier and more frequently in the MMF group (33%) compared with the CYC group (19%). Although this was a secondary outcome and the trial was not designed or powered to detect differences in relapse rate, this observation is consistent with the increase in early relapses observed with methotrexate compared with CYC,15 higher relapse risk with lower cumulative CYC exposure28 and the higher rate of relapse with MMF compared with AZA when used for maintenance therapy.29 While treatment with MMF may be associated with a higher risk of relapse compared with pulsed CYC, this increased risk may be acceptable to avoid the potential adverse effects of CYC particularly when the baseline risk of relapse is low (eg, in patients who are MPO-ANCA positive) or if rituximab is unavailable.
The use of MMF alongside standard dose glucocorticoids offers advantages over CYC in terms of fertility preservation for younger patients and potentially lower malignancy rates in elderly populations at greatest risk.30 Unlike rituximab (an approved alternative to CYC for severe AAV), MMF is an oral drug, has a short duration of action and, unlike methotrexate, can be used in moderate or severe renal disease and was not associated with slower time to remission compared with CYC.15 However, there were no differences in this study in the number of adverse events between the two groups.
Our trial has several notable strengths. It is the largest randomised trial in AAV to assess the use of MMF for remission induction. Patients were recruited from 21 countries, and the trial cohort was representative of other trial populations in AAV. This is the first randomised trial in AAV to include children, although the small number of paediatric participants (n=8) limits the inferences we might draw concerning relative efficacy of MMF in this population. The primary endpoint was achieved in one of four paediatric patients in both CYC and MMF groups and response rates were similar in the MMF and CYC groups in children. Compliance was a contributory factor to the lower remission rate in children, and because of the small sample size we have not drawn conclusions of efficacy in this subpopulation.
The strengths of our trial should be viewed against its limitations. The trial was not blinded, although the similar rates of glucocorticoid adherence and exposure, progressive disease, rescue therapy requirement, ANCA negativity and the rates of ESRD are reassuring. Treating clinicians were allowed to include plasma exchange or additional Solu-Medrol at entry; however, there were no differences in additional treatments used between the two groups. The short follow-up of 18 months in this study may have reduced the ability to detect the true effect on relapse and malignancy rates in the longer term. It should be noted in another study MMF was inferior to AZA for remission maintenance after CYC induction, with more relapses in the MMF group,.29 Following remission induction all patients in our trial received AZA and glucocorticoid maintenance therapy. There is limited evidence for using AZA as induction therapy in AAV. It has been used in addition to corticosteroids for newly diagnosed non-severe eosinophilic GPA, MPA or polyarteritis nodosa; however, the addition of AZA in these patients did not improve remission rates or reduce relapse.31
Since initiation of the trial, it has become common to use rituximab as an alternative to CYC induction therapy, which may question the use of MMF as an alternative induction therapy. However, rituximab is expensive and its use is restricted in many countries, for example, in New Zealand treatment of patients with MPO-ANCA vasculitis must first have failed with CYC or MMF14 prior to rituximab use. Alternative effective low-cost induction therapies may be required in some cases.
This study provides evidence that MMF is a potential alternative to CYC for remission induction in non-life-threatening AAV, particularly in patients with low predicted relapse risk, such as the elderly who are MPO positive. With increasing remission induction treatment options for AAV, stratified treatment approaches are indicated in order to optimise long-term outcomes.http://dx.doi.org/10.1136/annrheumdis-2018-214245
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Acknowledgments
We are very grateful to the trial adjudication committee for blinded data adjudication, and to Dr Pani Gopaluni and Dr Mark McClure for independent data adjudication. We are also grateful to Dr Afzal Chaudhry for the trial database design, all the trial investigators, subinvestigators, research nurses and all the patients who participated in this study. Support was also provided by the NIHR Cambridge Biomedical Research Centre. PB acknowledges support from the Great Ormond St Hospital Clinical Research Facility and NIHR Great Ormond St Biomedical Research Centre, and Great Ormond St Hospital Children’s Charity. We are grateful for the help provided by physicians from recruiting centres: Dr A Salama, University College London; Dr D Milford, Birmingham Children's Hospital; Dr A Short, University Hospital Coventry and Warwick; Dr D Kluth, University of Edinburgh; Dr R Luqmani, University of Oxford; Dr O Flossmann, Royal Berkshire Hospital; Dr J Stoves, Bradford Teaching Hospitals NHS Trust; Dr M Christian, Nottingham Children's Hospital; Dr E Baildam, Alder Hey Children's Hospital; Dr L Erwig, University of Aberdeen; Professor C Savage, University of Birmingham.
References
Supplementary materials
Lay summary
Disclaimer : This is a summary of a scientific article written by a medical professional (“the Original Article”). The Summary is written to assist non medically trained readers to understand general points of the Original Article. It is supplied “as is” without any warranty. You should note that the Original Article (and Summary) may not be fully relevant nor accurate as medical science is constantly changing and errors can occur. It is therefore very important that readers not rely on the content in the Summary and consult their medical professionals for all aspects of their health care and only rely on the Summary if directed to do so by their medical professional. Please view our full Website Terms and Conditions.
Copyright © 2019 BMJ Publishing Group Ltd & European League Against Rheumatism. Medical professionals may print copies for their and their patients and students non commercial use. Other individuals may print a single copy for their personal, non commercial use. For other uses please contact our Rights and Licensing Team.
Footnotes
LH and DJ are joint senior authors.
Handling editor Josef S Smolen
Collaborators Prof Alan D Salama, University College London; Dr David Milford, Birmingham Children's Hospital; Dr David Kluth, University of Edinburgh; Prof Raashid Luqmani, University of Oxford, Dr Oliver Flossmann, Royal Berkshire NHS Foundation Trust; Dr Andrew Short, University Hospitals Coventry and Warwick; Dr Eileen Baildam, Alder Hey Children's Hospital; Dr John Stoves, Bradford Teaching Hospitals NHS Foundation Trust; Dr Lars Erwig (was University of Aberdeen at time of study) now GlaxoSmith Kline Pharmaceuticals; Dr Martin Christian, Nottingham University Hospitals; Prof Caroline O Savage, (was University of Birmingham at time of study) now GlaxoSmith Kline Pharmaceuticals; all UK. All collaborators recruited at least 1 patient
Contributors All authors contributed to the design, conduct and/or analysis of the study. All authors reviewed the manuscript prior to submission and gave final approval.
Funding Sponsorship for this trial was provided by the Cambridge University Hospitals NHS Foundation Trust. Funding for this trial and the cost of the mycophenolate mofetil was provided in the form of a research grant from Vifor Pharma (previously Aspreva Pharmaceuticals). TFH is supported by NIHR 14/49/127, 16/167/120 and 17/27/11, and by the NIHR Cambridge Biomedical Research Centre. MCC and GEF acknowledge the support from Ministerio de Economía, Industria y Competitividad (SAF 14/57708-R and 17/88275-R) and Instituto de Salud Carlos III (PI 15/00092 cofunded by FEDER and Juan Rodés programme), respectively. The study was conducted within the Birmingham and Cambridge National Institute for Health Research (NIHR)/Wellcome Trust (WT) Clinical Research Facilities (CRF) at these sites.
Disclaimer The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health.
Competing interests RBJ: consulting for ChemoCentryx; academic secondment with GlaxoSmithKline 2011–2013. US: consulting for Genentech/Roche. PAM: consulting for Actelion, Alexion, Bristol Myers Squibb, ChemoCentryx, Genzyme/Sanofi, GlaxoSmithKline, Genentech/Roche, PrincipioBio; research support from Actelion, Bristol Myers Squibb, Celgene, ChemoCentryx, Genentech/Roche, GlaxoSmithKline. DJ: consulting for Alexion, ChemoCentryx, Genzyme/Sanofi, GlaxoSmithKline, Genentech/Roche and Takeda; research support from ChemoCentryx, Genentech/Roche, Genzyme/Sanofi, Medimmune and GlaxoSmithKline. LH: consulting for ChemoCentryx; honorarium from Roche. TFH: research support from GlaxoSmithKline, Otsuka and AstraZeneca.
Patient consent for publication Not required.
Ethics approval Oxfordshire Research Ethics Committee B (reference number 06/Q1605/120).
Provenance and peer review Not commissioned; externally peer reviewed.