Article Text

Abstract
Objective Immune-mediated inflammatory diseases (IMIDs) are heterogeneous and complex conditions with overlapping clinical symptoms and elevated familial aggregation, which suggests the existence of a shared genetic component. In order to identify this genetic background in a systematic fashion, we performed the first cross-disease genome-wide meta-analysis in systemic seropositive rheumatic diseases, namely, systemic sclerosis, systemic lupus erythematosus, rheumatoid arthritis and idiopathic inflammatory myopathies.
Methods We meta-analysed ~6.5 million single nucleotide polymorphisms in 11 678 cases and 19 704 non-affected controls of European descent populations. The functional roles of the associated variants were interrogated using publicly available databases.
Results Our analysis revealed five shared genome-wide significant independent loci that had not been previously associated with these diseases: NAB1, KPNA4-ARL14, DGQK, LIMK1 and PRR12. All of these loci are related with immune processes such as interferon and epidermal growth factor signalling, response to methotrexate, cytoskeleton dynamics and coagulation cascade. Remarkably, several of the associated loci are known key players in autoimmunity, which supports the validity of our results. All the associated variants showed significant functional enrichment in DNase hypersensitivity sites, chromatin states and histone marks in relevant immune cells, including shared expression quantitative trait loci. Additionally, our results were significantly enriched in drugs that are being tested for the treatment of the diseases under study.
Conclusions We have identified shared new risk loci with functional value across diseases and pinpoint new potential candidate loci that could be further investigated. Our results highlight the potential of drug repositioning among related systemic seropositive rheumatic IMIDs.
- gene polymorphism
- autoimmune diseases
- autoimmunity
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Systemic Seropositive Rheumatic diseases share symptoms, progressions, environmental risk factors, high rates of familial aggregation and susceptibility genes, pointing to a shared genetic architecture.
The assessment of a shared genetic component among these conditions has not been performed before in a systematic fashion.
What does this study add?
We identified 27 shared loci among systemic seropositive rheumatic immune-mediated inflammatory diseases, five of which were new. The remaining were known susceptibility genes in autoimmunity.
The associated variants were enriched in marks related with gene activation in immune cells and constitute shared expression quantitative trait loci.
How might this impact on clinical practice or future developments?
Most of these conditions do not have specific treatments, therefore, therapy repositioning among genetically related conditions could be feasible among them.
Introduction
Autoimmunity occurs when the mechanisms related to immune self-tolerance fail, leading to an inappropriate destruction of normal tissue by the immune system. Genetic factors play an important role in the development of more than 80 immune-mediated inflammatory diseases (IMIDs) identified so far.1 Comorbidity of these diseases, increased familial clustering and shared risk variants have been widely documented.2 However, to date, these shared loci have been identified by simple comparison between studies, and just recently they have been determined by rigorous and systematic analysis.3 In this sense, combining genome-wide association studies (GWAS) across several diseases has proven to be a very useful tool for the identification of new genetic risk variants simultaneously associated with several IMIDs and to expose shared pathways involved in the pathophysiology of these conditions.4–7 To date, two large studies combining several diseases were recently published following this strategy. One of them was a meta-GWAS across 10 paediatric autoimmune diseases with shared population-based controls that revealed new candidate loci with immunoregulatory functions.8 In the other study, the authors identified new shared associations by combining immunochip data across five chronic inflammatory diseases.9
Systemic seropositive rheumatological IMIDs, such as systemic sclerosis (SSc), systemic lupus erythematosus (SLE), rheumatoid arthritis (RA) and idiopathic inflammatory myopathies (IIM), are heterogeneous diseases of the connective tissue that share clinical and epidemiological manifestations as well as life-threatening complications.10 The common genetic component of these conditions has not been previously assessed systematically, although the overlap of associated genes is elevated when performing a pairwise comparison.8 Autoantibody production is the main feature of these diseases, comprising additionally a broad deregulation of the innate and adaptive immune response. However, the low prevalence of most of these diseases hinders the collection of large datasets that makes possible to attain sufficient statistical power. Therefore, our study aimed to combine previously published GWAS datasets—all from European descent populations—to identify shared genetic aetiologies among systemic seropositive rheumatological IMIDs in a systematic fashion.
Subjects and methods
Study population
A total of 12 132 affected subjects with four systemic seropositive rheumatic IMIDs (SSc, SLE, IIM and RA) and 23 260 controls were included in this study from previously published GWAS11–16 (online supplementary table S1).
Supplemental material
Data quality control and imputation
Unified quality control (QC) of the 18 case-control collections was conducted separately, based on stringent criteria using PLINK V.1.07.17 Given that related and/or duplicated subjects may have been recruited for different studies, genome-wide relatedness was assessed and one individual from each pair was removed. Samples with <95% of successfully called genotypes were removed.
Further, single nucleotide polymorphisms (SNPs) with genotyping call rate <98%, minor allele frequencies (MAF) <1% and deviating from Hardy-Weinberg equilibrium with a p<0.001 in the control group were removed. To control for possible population stratification, we performed principal component (PC) analysis using GCTA64 and R-base software under GNU Public license V.2.
Imputation of autosomal SNPs was conducted in the Michigan Imputation Server using Minimac3.18 The software SHAPEIT19 was used for haplotype reconstruction and the Haplotype Reference Consortium r1.1 was used as the reference population.20
Statistical analyses
Disease-specific association testing: association testing for allele dosages was performed by logistic Wald test using EPACTS software,21 adjusting by the first two or five PCs as appropriate to control for the genomic inflation factor in European population (λ<1.05) (online supplementary table S1). SNPs with a MAF≥1% and squared correlation (Rsq)≥0.3 were maintained in the analyses as suggested by the imputation software. Additionally, we calculated a concordance rate by comparing imputed and true genotypes.
Cross-phenotype meta-analysis: to identify shared loci, the summary-level statistics were meta-analysed using METASOFT.22 A fixed-effects model was applied for those SNPs without evidence of heterogeneity (Cochran’s Q test p value Q>0.05), and random-effects model was applied for SNPs displaying heterogeneity of effects between studies (Q≤0.05). Genome-wide significance was established at a p≤5×10−08. SNP independence was assessed with the software GCTA-COJO (online supplementary table S2).23 24 To annotate the independent signals, SNPnexus25 was used to the build 37 genomic coordinates.
Model search to identify the diseases contributing to the association: to identify the diseases most likely contributing to the association signals, we performed an exhaustive disease-subtype model search with the R statistical package ASSET.24 The contribution of a disease was considered if at least two independent case-control collections from the same disease were grouped with consistent effects.
Novelty of the variants: our independent SNP associations were classified into ‘known’ or ‘new’ associations based on the information retrieved from the NHGRI-EBI GWAS catalogue and the Phenopedia and Genopedia from HuGE Navigator.26
Functional enrichment analysis: in order to systematically characterise the functional, cellular and regulatory contribution of the associated variants, a non-parametric enrichment analysis implemented in GARFIELD was performed.27 Furthermore, the online tools HaploReg V.4.128 and the Genotype-Tissue Expression project (GTEx)29 were queried to determine whether any of the lead associated variants was an expression quantitative trait locus (eQTL). The online tool Capture HiC plotter was used to assess physical interactions between restriction fragments containing the variants and the promoter of genes in the three-dimensional nuclear space.30
Drug target enrichment analysis: the target genes of the eQTLs were used to model a protein-protein interaction (PPI) network using String V.10.31 These protein products were then used to query the OpenTargets Platform32 for drug targets. Moreover, this platform was used to search for drugs indicated or in different phases of drug development for the treatment of SSc, SLE, IIM and RA. The Fisher’s exact test was used to calculate if the results of the meta-analysis were significantly enriched in pharmacologically active drug targets.
Additional details of the Methods section are available in the online supplementary methods.
Results
Cross-phenotype meta-analysis and disease contribution
Following sample QC and imputation, a total of 11 678 cases and 19 704 non-overlapping controls were included in the genome-wide meta-analysis of 6 450 125 SNPs across the four diseases. The mean concordance rate among imputed and true genotypes was 0.999±0.0003. The final λ showed minimal evidence of population stratification in the meta-analysis (λ=1.025). Moreover, we calculated λ1000 with consistent results (λ1000=1.025). Summary of sample/variant QC and QQ plots are shown in online supplementary table S1 and figure S1, respectively.
The global meta-analysis revealed 42 non-hla significantly associated loci. Subsequent conditional analyses showed that 27 SNPs were independent (figure 1 and online supplementary figure S2). Sixteen variants were meta-analysed under a fixed effects model, whereas 11 with random effects based on study heterogeneity.

Meta-analysis results for the four systemic IMIDs. The Manhattan plot displays the −log10 transformed p values (y-axis) by position on each chromosome (x-axis). The red line depicts the genome-wide significance threshold (p=5×10−8). A total of 26 SNPs were independently associated with at least 2 systemic IMIDs. Most of the signals map to known susceptibility loci in autoimmunity (eg, PTPN22, STAT4, TNPO3, FAM167A-BLK) and five loci have never been reported before. IMIDs, immune-mediated inflammatory diseases.
To comprehensively explore the combinations of diseases contributing to the associations we applied a subset-based meta-analysis implemented in ASSET.24 Our model search yielded 26 SNPs associated with at least two IMIDs (table 1). All of these variants were imputed in at least one dataset.
Among these 26 associations, we found several key players in autoimmunity; interestingly, 10 of these associations (38%) have never been reported before for SSc, 8 (31%) for SLE and RA, respectively and 20 (77%) for IIM. Remarkably, five SNPs have not been reported previously for any of the diseases under study and thus constitute new shared risk loci in systemic seropositive rheumatic IMIDs (table 1). Among these five new associations we found the SNP rs744600 in the 3’ region of the NGFI-A binding protein 1 (NAB1) (OR for the T allele 0.88, 95% CI 0.85 to 0.92), p=7.07×10−11) and the intronic SNP rs13101828 mapping in the gene Diacylglycerol kinase theta (DGKQ) (OR for the G allele 1.11, 95% CI 1.07 to 1.16, p=1.32×10−08). Of note, both genes have been previously associated with a chronic autoimmune liver disease.33 34 The intergenic SNP rs112846137, maps between the genes Karyopherin subunit alpha 4 (KPNA4) and the ADP ribosylation factor like GTPase 14 (ARL14) (OR for the T allele 1.29, 95% CI 1.07 to 1.56, p=1.42×10−08). Interestingly, the gene ARL14 showed a suggestive association in a pharmacogenomic GWAS of response to methotrexate in patients with RA.35 In addition, we observed the associated SNP rs193107685 located in the 3’ region of the LIM domain kinase 1 (LIMK1) gene (OR for the C allele 1.52, 95% CI 1.27 to 1.83, p=3.81×10−09). The protein encoded by this gene regulates actin polymerisation, a critical process in the activation of T cells.36 Finally, the SNP rs76246107 is located in an intron of the gene Proline rich 12 (PRR12) (OR for the G allele 1.28, 95% CI 1.14 to 1.43, p=3.36×10−08), which was associated with fibrinogen concentration,37 and is an active regulator of the inflammatory response.38
Twenty-six independent variants associated at a genome-wide significance level (p<5×10−8) in the meta-analysis
Associated loci and their functional enrichment on regulatory elements
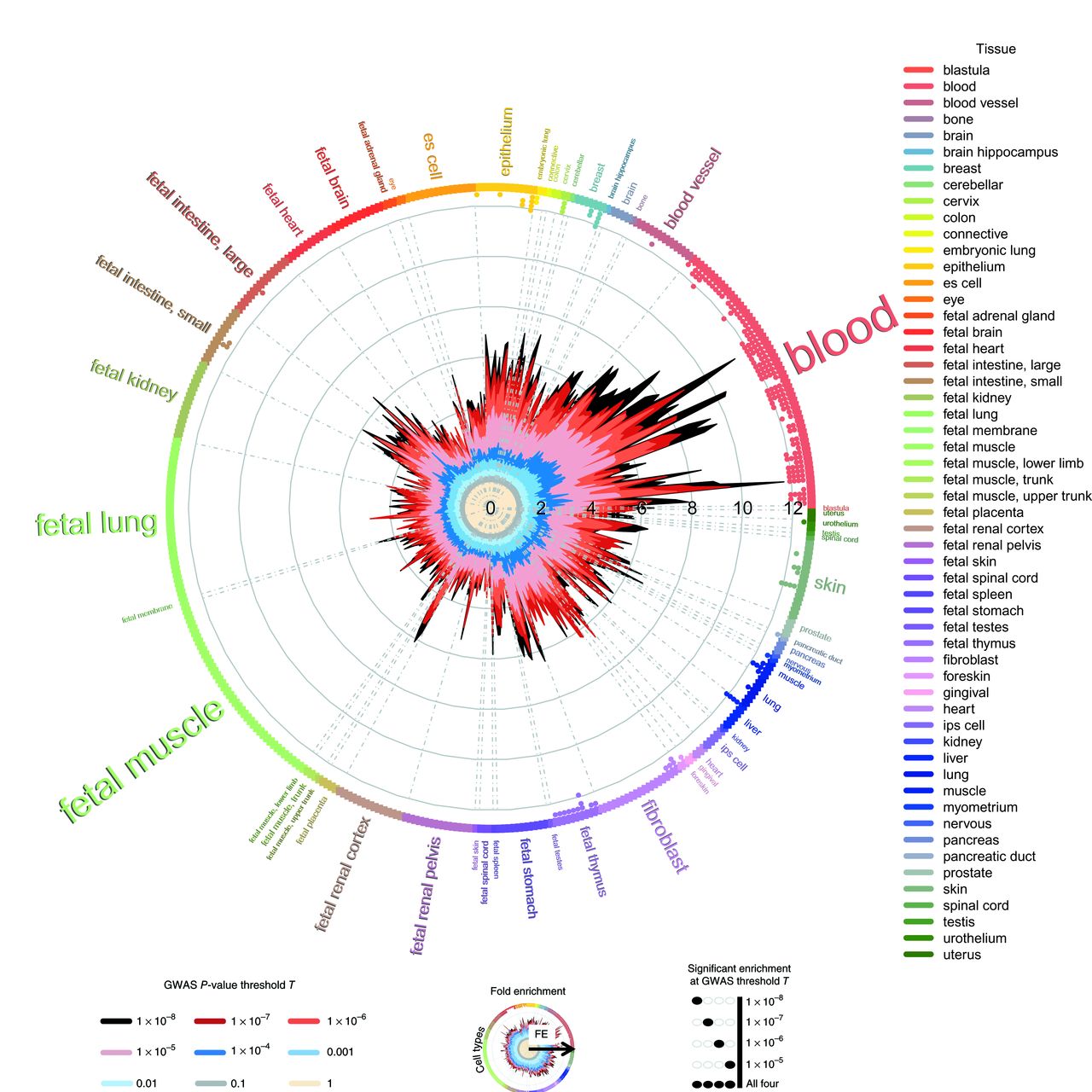
To assess whether the associated variants lie in coding and non-coding regulatory and cell-type-specific elements of the genome, we performed an enrichment analysis with GARFIELD.39 The results obtained showed marked enrichment patterns mainly in blood cells and skin cells, with 247 significant enrichments (p≤5×10−05) (online supplementary figure S3 and table S3). Table 2 summarises the main enrichment results. We found that the majority of associated variants were enriched in DNase I hypersensitivity site hotspots in blood, as depicted in figure 2. This functional category included a repertoire of cells from the immune system, such as B-lymphocytes (fold enrichment (FE)=11.68, empirical p (pemp) <1×10−05), T-lymphocytes (FE=10.42, pemp <1×10−05), including T helper cells (FE=7.81, pemp <1×10−05), T CD8+ (FE=7.61, pemp <1×10−05), natural killer cells (FE=11.36, pemp <1×10−05) and monocytes (FE=8.99, pemp <1×10−05). In line with this enrichment, disease-associated SNPs were enriched in enhancers (FE=14.99, pemp <1×10−05), within TSS (FE=14.87, pemp <1×10−05) and on transcription factor binding sites (FE=12.20, pemp <1×10−05) in the B-lymphocyte cell line GM12878. Additionally, the highest enrichment was observed in the histone modification H3K9ac (FE=14.02, pemp <1×10−05) and H3K27ac (FE=10.81, pemp <1×10−05) in the B-lymphocyte cell line, which are positively associated with gene activation. Although these modifications are increased in the promoters of active genes, the latter has been shown to be associated with active enhancers.40 Moreover, enrichment was observed in H3K4me1,2,3 sites, which usually surround TSS and are also positively correlated with gene expression.40
Summary of the most enriched functional annotations for the SNPs associated in the meta-analysis at a genome-wide significance threshold (p<5×10−8)
{kind=link}
{kind=link}
GARFIELD functional enrichment analyses in DHS hotspots. The wheel plot shows functional enrichment in systemic IMiDs within DHS hotspot regions in encode and roadmap epigenomics. The radial axis depicts the FE calculated at different meta-analysis p value thresholds. The font size is proportional to the number of cell types from the tissue, mainly enriched in blood cell types including a repertoire of immune cell lines. DHS, DNase I hypersensitivity site; FE, fold enrichment; IMIDs, immune-mediated inflammatory diseases.
Expression quantitative trait loci (eQTL) and associated variants
In silico analysis of eQTLs revealed the role of 16 of the lead SNPs as eQTLs in whole blood, lymphoblastoid cell lines, transformed lymphocytes, skeletal muscle and transformed fibroblasts derived from European individuals from HaploReg V.4.128 (table 3 and online supplementary table S4). Focusing on new associated variants, the SNP rs744600 modifies NAB1 gene expression in lymphoblastoid cell lines (p=1.30×10−34), whereas the T allele increases HIBCH expression in skeletal muscles (p=8.09×10−07). The G allele of rs13101828 increases DGKQ expression in whole blood (p=3.29×10−45), lymphocytes (p=5.23×10−19), fibroblasts (p=4.44×10−06), lung cells (p=8.42×10−28) and several other tissues. The A allele of rs76246107 can reduce ALDH16A1 expression in lung cells (p=6.45×10−06), and the protein encoded by this gene is involved in oxidoreductase activity. Reassuringly, 14 of the 16 (87%) reported eQTLs showed a physical interaction between the SNP and the promoter of 15 of the genes affected by the eQTLs (table 3), as suggested by Capture HiC (C-HiC) data (online supplementary table S5). These independent evidences propose a mechanistic approach to understand the modulation of gene expression.
Summary of the eQTL results in European samples for the SNPs independently associated in the meta-analysis
Drug target enrichment analysis
Genetic associations have the potential to improve the rates of success in the development of new therapies.41 We assessed if the protein-products from disease associated eQTLs and their direct PPI partners were enriched with pharmacologically active targets (online supplementary tables S6 and S7). We identified as eQTLs and PPIs 608 proteins for SSc, 630 for SLE, 632 for IIM and 413 for RA, based on data on drugs at any stage of development collected from the Open Targets Platform (online supplementary table S8).32 Using this information, we found for SSc that 23 out of 73 (32%) proteins are targeted by drugs being studied for the disease (OR=16.80, p=1.41×10−18). Similarly, 7 out of 25 (28%) proteins related to IIM and 13 out of 146 (9%) proteins related to SLE are addressed by drugs in consideration for IIM and SLE (OR=13.40, p=4.62×10−06, OR=3.38, p=2.85×10−04, respectively) (online supplementary table S9).
Discussion
In the present study, we identified five unreported shared loci associated with systemic seropositive rheumatic IMIDs. This is the first large-scale meta-analysis, including more than 11 000 cases and 19 000 non-overlapping controls aiming to improve our knowledge regarding the genetic resemblances among these conditions.
Our results show that 85% of the associated variants were shared by at least three diseases. Interestingly, for several known RA susceptibility loci, the contribution of RA was limited. In this case, most of the associated variants were independent to the ones previously reported. Among the new associated SNPs, the signals mapping to NAB1, DGKQ and KPNA4-ARL14 were associated to all of the diseases under study. NAB proteins are known to interact with early growth response family members and act as corepressors induced by type I interferons (IFN).42 The ‘IFN signature’ has been previously described in these diseases.43–46 Interestingly, two IFN regulatory factors—IRF5 and IRF8—previously associated to the conditions under study, were associated in the meta-analysis. Additionally, the associated SNP is an eQTL in lymphoblastoid cell line, which evidences its role in disease pathogenesis. The DGKQ protein mediates cell signal transduction and can indirectly enhance the epidermal growth factor receptor signalling activity.47 This pathway regulates cell proliferation and migration, and its expression is augmented in the vasculature of patients with SSc with pulmonary involvement.48 Moreover, the risk allele was associated with an increased expression of the gene in lymphocytes, fibroblasts and lung. In the same line, this gene was associated with Sjögren's syndrome, a related connective tissue disease.49 The protein encoded by the gene ARL14 is a GTPase involved in the recruitment of MHC class II containing vesicles and control the movement of dendritic cells (DCs) along the actin cytoskeleton.50 The protein LIMK1 regulates many actin-dependent processes, including the assembly of the immune synapse between T cells and antigen presenting cells, an expected biological process involved in seropositive IMIDs. Remarkably, rs193107685 and rs112846137 interact physically with the promoters of the genes LIMK1 and ARL14, respectively, in DCs (online supplementary figure S1). The gene PRR12 has been previously associated with fibrinogen concentrations.37 Fibrinogen is considered a high-risk marker for vascular inflammatory diseases and is considered an accurate predictor of cardiovascular diseases.38 51 Moreover, this molecule is an active player in the coagulation cascade, responsible for the spontaneous formation of fibrin fibrils. Cardiovascular events and fibrosis are the most life-threatening complications described in SSc, IIM and SLE.52–54
The associated SNPs are highly enriched in functional categories in B and T cells, natural killer and monocytes, highlighting the relevance of these cells in systemic seropositive rheumatic IMIDs. Beyond whole blood, the skin is the other tissue with significant functional categories, which is not surprising given the nature of these connective tissue diseases. Moreover, epithelial cells could transdifferentiate into mesenchymal cells and eventually contribute in fibrotic processes.55 Moreover, patients with SSc are usually stratified according to the extent of skin involvement.43 On the other hand, the histone modifications observed are consistent with the ones reported in previous studies, where histone hyperacetylation have been described in synovial tissues in RA, in B cells in SSc and in CD4+T cells in SLE.40 Finally, the independent associated SNPs have significant eQTLs in relevant tissues (table 3) and in silico data from promoter capture HiC experiments showed the potential mechanisms in which most eQTLs modulate gene expression. Interestingly, all new associated SNPs interact with the promoters of surrounding genes, suggesting them as putative candidates with a role in the pathophysiology of these conditions (online supplementary tables S4 and S5).
The prevalence of SSc, SLE and IIM is low and there are no specific treatments for these diseases in comparison with RA; therefore, given our current knowledge on the use of genetic findings in drug target validation and drug repurposing, we evaluated if drugs currently indicated for RA had the potential to be used in any of the other IMIDs under study. Our meta-analysis revealed that ten loci overlap with known RA risk genes. For instance, the gene-product of TYK2 is targeted directly by Tofacitinib, which inhibits janus kinases (https://www.drugbank.ca/drugs/DB08895) or indirectly through the interleukin 6 (IL-6) family signalling pathway by targeting the IL6 receptor with Tocilizumab (https://www.drugbank.ca/drugs/DB06273). Both drugs are currently indicated for patients with moderate to severe RA who respond poorly to disease-modifying antirheumatic drugs. As TYK2 is associated with SSc, SLE and IIM, it is a good candidate for therapy repositioning in these diseases. As a proof of concept, Tofacitinib is currently on trial for SLE (clinical trial identifier NCT02535689), SSc (NCT03274076) and Dermatomyositis (NCT03002649). Overall, we found that five of the loci identified in our meta-analysis interact with 17 genes that are considered drug targets, six of which are used for the treatment of these diseases (table 4). Another interesting candidate for drug repurposing is Imatinib, a kinase inhibitor that targets ABL1, which interacts with the gene product of BLK, a known locus associated with SSc and RA (table 4). Imatinib is currently being tested for SSc (NCT00555581) and RA (NCT00154336).
Summary of the plausible target gene products with drug indications in systemic IMIDs
As compared with previous cross-phenotype studies of autoimmune diseases, our study has the strength of analysing systemic seropositive rheumatic diseases, which is a consistent clinical phenotype than in the diseases investigated previously, where mixed seropositive and seronegative diseases were analysed, and combining systemic and organ-specific diseases.8 9 The study of a more homogenous phenotype allowed us to determine that the type I IFN signalling pathway and its regulation play a more prominent role in these conditions than in others, based on the associations observed in NAB1, TYK2, PTPN11, IRF5 and IRF8. Additionally, we performed a genome-wide scan to identify shared genetic aetiologies, as opposed to the study performed by Ellinghaus et al whose analyses were limited to the 186 autoimmune disease-associated loci implemented in the Immunochip platform. The study performed by Li et al —which was also a meta-analysis of GWAS data—was focused on paediatric autoimmune diseases, whereas our study was on a new combination of diseases in adult population.
In summary, this is the first study to investigate shared common genetic variation in four systemic seropositive rheumatic IMIDs in adults. We identified 26 genome-wide significant independent loci associated with at least two diseases, of which five loci had not been reported before. The shared risk variants and their likely target genes are functionally enriched in relevant immune cells and significantly enriched in drug targets, indicating that it may assist drug repositioning among genetically related diseases based on genomics data.
Acknowledgments
We would like to thank Sofia Vargas, Sonia García, and Gema Robledo for their excellent technical assistance, as well as Dr Carlos Flores, Dr Maria Pino-Yanes, Dr Elaine Remmers and Dr Doug Bell for their outstanding advice. Additionally, we would like to thank all the patients and healthy controls for their essential collaboration and the MYOGEN consortium for the provision of data. The results reported in this study were presented in the Genomics of Common Diseases 2017 conference and in EULAR 2018 (Acosta-Herrera M, Kerick M, González-Serna D et al. Ann Rheum Dis 2018; 77 Supp 2).
References
Footnotes
MA-H and MK are joint first authors.
MA-H and JM contributed equally.
Handling editor Josef S Smolen
Collaborators Myositis Genetics Consortium: (1) Frederick W Miller; (2) Wei Chen; (1) Terrance P O’Hanlon; (3) Robert G Cooper; (4) Jiri Vencovsky; (1) Lisa G Rider; (5) Katalin Danko; (6) Lucy R Wedderburn; (7) Ingrid E. Lundberg; (8) Lauren M Pachman; (9) Ann M. Reed; (9) Steven R Ytterberg; (10) Albert Selva-O’Callaghan; (11) Timothy R Radstake; (12) David A Isenberg; (13) Hector Chinoy; (14) William E R Ollier; (2) Paul Scheet; (2) Bo Peng; (15) Annette Lee; (14) Janine A Lamb; (16) Christopher I Amos; (17) Christopher Denton; (18) David Hilton-Jones; (19) Paul H Plotz; (20) Hemlata Varsani. (1) National Institute of Environmental Health Sciences, National Institutes of Health, Bethesda, MD, USA; (2) MD Anderson Cancer Center, Houston, Texas, USA; (3) MRC/ARUK Institute for Ageing and Chronic Disease, University of Liverpool, UK; (4) Institute of Rheumatology, Charles University, Prague, Czech Republic; (5) 3rd Department of Internal Medicine, Division of Immunology University of Debrecen, Debrecen, Hungary; (6) Institute of Child Health, University College London, London, UK; (7) Rheumatology Unit, Department of Medicine, Karolinska University Hospital, Solna, Karolinska Institutet, Stockholm, Sweden; (8) Department of Pediatric Rheumatology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA; (9) Mayo Clinic, Rochester, Minnesota, USA; (10) Vall d'Hebron General Hospital, Barcelona, Spain; (11) Department of Rheumatology and Clinical Immunology, Laboratory for Translational Immunology, Utrecht University Medical Center; and Nijmegen Center for Molecular Life Sciences, Nijmegen, The Netherlands; (12) Division of Medicine, University College London, London, UK; (13) The National Institute for Health Research Manchester Musculoskeletal Biomedical Research Unit, Centre for Musculoskeletal Research, University of Manchester, Manchester, UK; (14) Centre for Integrated Genomic Medical Research, Manchester Academic Health Science Centre, University of Manchester, Manchester, UK; (15) Robert S. Boas Center for Genomics and Human Genetics, Feinstein Institute for Medical Research, Manhasset, New York, USA; (16) Department of Community and Family Medicine, Dartmouth College, Hanover, NH, USA; (17) Centre for Rheumatology, Royal Free Hospital, London, UK; (18) Nuffield Department of Clinical Neurosciences, University of Oxford, Oxford, UK; (19) National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health, Bethesda, Maryland, USA; (20) University College London, London, UK.
Scleroderma Genetics Consortium: Timothy R.D.J. Radstake,1Olga Gorlova,2 Blanca Rueda,3 Jose-Ezequiel Martin,3Behrooz Z. Alizadeh,4 Rogelio Palomino-Morales,3Marieke J. Coenen,5 Madelon C. Vonk,1 Alexandre E.Voskuyl,6 Annemie J. Scheurwegh,7 Jasper C. Broen,1Piet L.C.M. van Riel,1 Ruben van ‘t Slot,4 AnnetItaliaander,4 Roel A. Ophoff,4,8 Gabriela Riemekasten,9Nico Hunzelmann,10 Carmen P. Simeon,11 NorbertoOrtego-Centeno,12 Miguel A. González-Gay,13 María F.González-Escribano,14 Paolo Airo,15 Jaap van Laar,16Ariane Herrick,17 Roger Hesselstrand,18 Vanessa Smith,19Filip de Keyser,19 Fredric Houssiau,20 Meng May Chee,21Rajan Madhok,21 Paul Shiels,21 Rene Westhovens,22Alexander Kreuter,23 Hans Kiener,24 Elfride de Baere,25Torsten Witte,26 Lars Klareskog,27 Lorenzo Beretta,28Rafaella Scorza,28 Benedicte A. Lie,29 Anna-MariaHoffman-Vold,30 Patricia Carreira,31 John Varga,32Monique Hinchcliff,32 Annette T. Lee,32 Jun Ying,2Younghun Han,2 Shih-Feng Weng,2 Fredrick M. Wigley,33Laura Hummers,33 J. Lee Nelson,34 Sandeep K. Agarwal,35Shervin Assassi,35 Pravitt Gourh,35 Filemon K. Tan,35Bobby P.C. Koeleman,4 Frank C Arnett.35
1Radboud University NijmegenMedical Center, Department of Rheumatology, The Netherlands; 2Departmentof Epidemiology, M.D. Anderson Cancer Center, Houston, TX, USA; 3Instituto de Parasitologíay Biomedicina López-Neyra, CSIC, Granada, Spain; 4Department of MedicalGenetics, University Medical Center Utrecht, The Netherlands; 5RadboudUniversity Nijmegen Medical Center, Department of Human Genetics, TheNetherlands; 6Department of Rheumatology, VU University MedicalCentre, Netherlands; 7Department of Rheumatology, University ofLeiden, The Netherlands; 8UCLA Center for Neurobehavioral Genetics,Los Angeles, California; 9Department of Rheumatology and ClinicalImmunology, Charité University Hospital, Berlin, Germany; 10Departmentof Dermatology, University of Cologne, Germany; 11Servicio deMedicina Interna, Hospital Valle de Hebron, Barcelona, Spain; 12Serviciode Medicina Interna, Hospital Clínico Universitario, Granada, Spain; 13Serviciode Reumatología, Hospital Marqués de Valdecilla, Santander, Spain; 14Serviciode Inmunología, Hospital Virgen del Rocío, Sevilla, Spain; 15Universityof Brecia, Italy; 16University of Newcastle, United Kingdom; 17Universityof Manchester, United Kingdom; 18University of Lund, Sweden; 19Universityof Ghent, Belgium; 20University of Leuven, Belgium; 21Universityof Glasgow, United Kingdom; 22University of Antwerpen, Belgium; 23RuhrUniversity of Bochum, Germany; 24University of Vienna, Austria; 25Departmentof Genetics, University of Ghent, Belgium; 26University of Hannover,Hannover, Germany; 27Karolinska Institute, Stockholm, Sweden; 28Universityof Milan, Italy; 29Institute of Immunology, Rikshospitalet, OsloUniversity Hospital, Oslo, Norway; 30Department of Rheumatology,Rikshospitalet, Oslo University Hospital, Oslo, Norway; 31Hospital12 de Octubre, Madrid; 32Feinstein Institute of Medical Research,Manhasset, NY, USA; 33Johns Hopkins University Medical Center,Baltimore, MD, USA; 34Fred Hutchinson Cancer Research Center,Seattle, WA, USA; 35The University of Texas Health ScienceCenter-Houston, Houston, TX, USA.
Contributors Data providers: FWM, WC, TPO, RGC, JV, LGR, KD, LRW, IEL, LMP, AMR, SRY, ASO, TRR, DAI, HC, WERO, PS, BP, AL,JAL, CIA, CD, DHJ, PP, HV, on behalf of the Myositis Genetics Consortium; OG, BR, JEM, BZA, RPM, MJC, MCV, AEV, AJS, JCB, PLCMR, RS, AI, RAO, GR, NH, CPS, NOC, MAGG, MGE, PA, JVL, AH, RH, VS, FDK, FH, MMC, RM, PS, RW, AK, HK, EDB, TW, LK, LB, RS, BAL, AMHV, PC, JV, MH, ATL, JY, YH, SFW, FMW, LH, JLN, SKA, SA, PG, FKT, BPCK, FCA, on behalf of the Scleroderma Genetics Consortium; QC and imputation in the contributing studies: MAH, MK, DGS; Functional and drug enrichment analysis: MAH, MK; Meta-analysis, tables and figures: MAH, MK, DGS; Drafting and approved version of the manuscript: MAH, MK, DGS,CW, AF, PKG, LP, JW, TV, MEAR, MDM, JM, FWM, WC,TPO, RGC, JV, LGR, KD, LRW, IEL, LMP, AMR, SRY, ASO, TRR, DAI, HC, WERO, PS, BP, AL, JAL, CIA, CD, DHJ, PP, HV; Study design and management: MAH, MK, JM.
Funding Funded by EU/EFPIA Innovative Medicines Initiative Joint Undertaking PRECISESADS (115565), The Spanish Ministry of Economy Industry and Competitiveness (SAF2015-66761-P), The Regional Ministry of Innovation, Science and Technologies of the Andalusian Regional Government (P12-BIO-1395) and Juan de la Cierva fellowship (FJCI-2015-24028). This research was supported in part by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval This study was conducted using available data included in previously published GWAS (online supplementary references 1–6).
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Summary statistics of the global meta-analysis generated and analysed in the current study are available from the corresponding author on reasonable request.