Article Text

Abstract
While biologic disease-modifying antirheumatic drugs (bDMARDs) have transformed outcomes of people with rheumatoid arthritis (RA), a proportion of patients are refractory to multiple bDMARDs. Definitions of refractory RA thus far have been arbitrary, and outcome data and impact of such cohorts remain limited. Extrapolation from randomised controlled trial and some real-life data suggest approximately 20% progress onto a third bDMARD with a more modest proportion failing additional bDMARDs. This viewpoint discusses an opinion of refractory RA disease and proposes key principles to accurately identify refractory cohorts. These include demonstrating presence of persistent inflammation despite multiple therapies and acknowledging development of antidrug antibody. Potential basis of refractory disease is summarised, and suggestions for an initial approach in the future evaluation of refractory disease are offered. Specific investigation of refractory RA disease is necessary to inform the clinical need and provide a basis for robust investigation of underlying mechanisms.
- rheumatoid arthritis
- dmards (biologic)
- treatment
Statistics from Altmetric.com
Background
While targeted therapies have transformed the management of rheumatoid arthritis (RA), refractory disease to multiple biologic disease-modifying antirheumatic drugs (bDMARDs) presents a significant clinical challenge. Extrapolating from randomised controlled trial (RCT) data (with low hurdle response endpoints), approximately 40% failure to a first bDMARD1 and another 40% on a second bDMARD,2–4 implies almost 20% progress to a third bDMARD. Disease progression and impact in such a multi-bDMARD cohort is not clear, a point highlighted by historical studies illustrating lack of structural progression in tumour necrosis factor inhibitor (TNFi)-treated cohorts.5 Our understanding of refractory RA disease thus remains limited.
This viewpoint focuses on multi-bDMARD inefficacy and proposes an approach to identify true (intrinsic) refractory RA disease (distinct from antidrug antibody (ADA)-mediated non-response) and possible underlying mechanisms. In the absence of a dedicated evidence base to refer to, this viewpoint reflects an opinion to highlight the need for and inform future initiatives.
Setting the scene
Refractory disease is broadly assumed to imply resistance/refractoriness of multiple agents, more than might be considered ‘normal’ or ‘reasonable’ for the specific disease. Prior to the introduction of bDMARDs, refractory RA denoted multiple conventional synthetic DMARD failure,6 although methotrexate (MTX)-inadequate response (IR) RCTs have historically not mandated failure to optimal dose MTX. With escalation to TNFi (as the first available bDMARD class), a subsequent (multiple) TNFi-failure cohort emerged. We now observe a cohort that has failed several bDMARD classes. The only registry data in this field report 5% failing at least third bDMARD class due to inefficacy and/or toxicity.7 RCTs typically demonstrate 50%–30%–10% ACR 20, 50 and 70 responses, respectively, in TNFi-IR studies.2–4 Thus, the vast proportion (over 70%) of patients on a second bDMARD class fail to actually derive a meaningful clinical response.
Identifying refractory RA disease: should necessarily mean refractory inflammation
Targeted agents are designed a priori to interfere with key mediators of inflammation and thus suppress synovial inflammation, the primary site of pathology and driver of joint damage.8 Thus, the assessment of an individual with RA refractory to a single and/or multiple DMARDs should necessarily mean presence of persistent (proven) inflammation, be it local synovitis and/or systemic inflammation.
Recognising discordance between measured disease activity and pathology
While clinical tools such as DAS28 are well-established validated surrogate measures of synovitis and employed for response assessment,9 limitations in this are well-recognised. Discordance is observed between clinically judged disease activity and validated clinical tools, the latter sometimes disproportionately driven by the more subjective components.10 11 Secondary damage and osteoarthritis associated with increasing disease duration as well as (poorly understood) chronic pain states are recognised to distort measured active disease state and thus could reasonably drive apparent ‘refractory’ drug profiles. If indicated, assessment of inflammation can be strengthened by imaging, with presence of power Doppler ultrasound, a credible measure that has been linked to damage.12
Identifying pharmacokinetic drivers and intrinsic refractory disease
Non-response is typically categorised as primary or secondary non-response based on whether an initial (usually defined as week 12–16) response to an intervention is observed or not.13–15 Incorrect targeting (ie, a mismatch between key disease mediator(s) and drug target) is a principal concept in the investigation of drug non-response (discussed later). Primary non-response has been presumed to be indicative of this. This suggestion of intrinsic disease resistance contrasts with pharmacokinetic factors in which drug is on target, but ADA-drug immune complexes lead to abrogation of pharmacological activity of the drug and/or enhanced drug clearance. This phenomenon is clinically relevant; it can be measured and potentially circumvented using alternative within-class bDMARD.15 16 ADA, however, tend to be considered in the context of secondary resistance but could conceivably develop in the earliest stages of treatment exposure.16 17 The conventional approach of using timing of non-response as a surrogate for incorrect targeting or ADA thus remains an assumption. Instead, an unbiased approach to demonstrate the basis of non-response would be more accurate clinically meaningful.
Mechanisms underlying refractory RA disease
No studies have specifically investigated the biological basis of multi-bDMARD refractoriness. Efficacy of non-TNF-targeted therapies in TNFi refractory cohorts and post-hoc analyses4 is used to support the assertion of incorrect drug targeting (or pathological accuracy) as the basis for individual refractory drug response. Comparative trials such as the ‘ROC’ trial18 in which non-TNF bDMARD class was superior to alternative TNFi in individuals with first TNFi lack of efficacy further suggests this interpretation (although limited by the non-TNFi arm comprising three different bDMARD classes). TNFi to TNFi switch however has been demonstrated to be effective,19 and the theory of incorrect targeting also does not necessarily play out in clinical practice where we often see strikingly opposite primary responses with within-class (TNFi) cycling.20 21
Furthermore, experimental investigations have not yet validated this concept.22 Within the elegantly described biological paradigm23 underpinned by clinical observation of a cytokine network that is host to key druggable nodes (such as TNF, IL-6 and GM-CSF), the inter-relationship of such cytokine nodes (does response to TNFi implicitly indicate TNF-driven disease not amenable to IL-6 targeting?), whether ‘adaptation’ to alternative key nodes or mechanisms may occur, and whether and how multi-bDMARD refractory RA fits in this concept, is unclear. Figure 1 further summarises the possible pathophysiological basis of refractory disease (a detailed appraisal of which is outside the scope of this viewpoint).
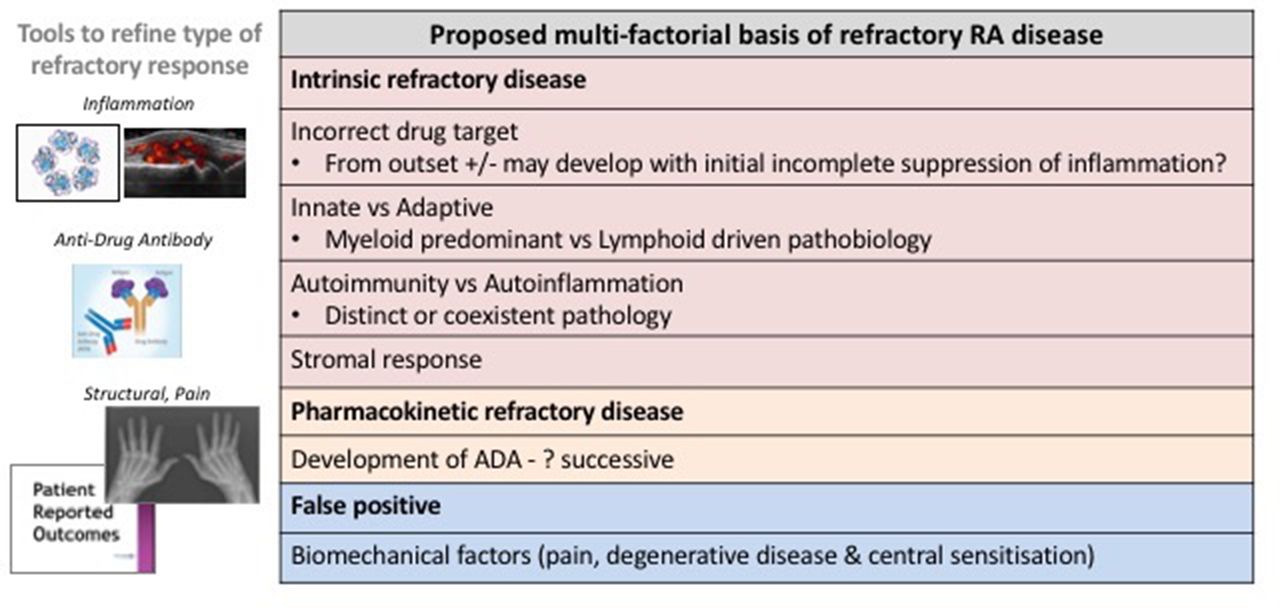
Refractory disease that is not as a consequence of ADA and/or biomechanical factors may represent disease subgroups with distinct immunopathological drivers. Different cytokine/cell pathway targets and associations between synovial tissue pathobiological subtype, associated genes and response to bDMARD have been suggested.28 The role of both innate and adaptive drivers of disease, and observation of autoinflammatory phenotype with/without coexistent typical autoantibody-mediated disease29 provides an additional basis for refractory subgroups. The relevance of the stromal response and effect of FLS-derived cytokines in driving persistence of synovitis in refractory RA disease might also be particularly relevant.30 ADA, antidrug antibody; bDMARD, biologic disease-modifying antirheumatic drug; FLS, fibroblast-like synoviocytes; RA, rheumatoid arthritis.
Research agenda
As a starting point, future research in multi-drug refractory RA disease requires consistent definitions and criteria.
Overarching definition of refractory RA disease
Similar to that employed in cancer therapy, refractory RA disease could on a generic level be defined as:
resistance to multiple therapeutic drugs with different structures and mechanisms of action.
Following optimal dose MTX inefficacy, the number of prior bDMARD an individual’s RA disease needs to be refractory to before classified as multi-drug refractory disease is not implicitly clear. Multiple within-class bDMARD resistance (as with TNFi cycling) would not seem compatible with refractory RA disease. With current bDMARD classes comprising two broad mechanisms (anticytokine and cell-targeted agents), one could in the first instance suggest the following:
failure of at least one anticytokine (TNF and/or IL-6 directed) and one cell-targeted (B cell depletion and/or T cell costimulation blockade) bDMARD.
The advent of Janus kinase inhibitors (JAKi), the first targeted synthetic molecule, adds another layer of complexity to the above definition that in time may incorporate failure to a targeted synthetic therapy.
Evaluating ADA and non-inflammatory drivers of refractory RA
Identifying ADA and non-inflammatory pathologies in clinical studies are necessary to be able to classify refractory response. Central to this is confirming persistent inflammation (synovitis and/or systemic), distinct from solely clinically relevant biomechanical and degenerative drivers, so that we do not only include a surrogate for longer disease duration and damage.24 Ultrasound imaging and presence of power Doppler is appropriate if/when clinical assessment is not clear.25 The presence or absence of ADA can provide further clarification such that refractory response can be stratified into the following groups (illustrated in figure 2 and also applied to categorisation of successive refractory drug outcomes):

{kind=link}
{kind=link}
Categorisation of individual and successive refractory drug response.* *Disease activity status may be verified with additional use of imaging such as ultrasound to confirm presence/absence of synovial inflammation. ADA, antidrug antibody; PK, pharmacokinetic; RA, rheumatoid arthritis.
intrinsic refractory: persistent inflammation, no ADA (with/without secondary damage)
pharmacokinetic refractory: persistent inflammation with ADA
false refractory: absence of inflammation; other (biomechanical±degenerative) drivers.
Coexistence of incorrect targeting with ADA is theoretically possible, but perhaps somewhat academic as the former could not be overcome without bDMARD class switch.
Minimum clinical target to determine refractory disease
Building on the treat to target principles of RA management,26 a minimum target tailored to the individual should be set that if not achieved would determine refractory disease state. Moderate disease activity (as measured by a validated scoring tool) may be the most appropriate in a large proportion (taking into account prevalent populations that may have accrued extensive secondary consequences of disease). For newly diagnosed patients, achievement of clinical target within 6 months of commencing conventional synthetic DMARD is advocated. Applying similar principles, intrinsic failure of a minimum of two classes of bDMARD would be reached within 18–24 months, providing an initial indication of time course in the development of multi-bDMARD refractory patients.
Investigating the biological relationship between synovial inflammation and pain
Observations from the more recent RCTs of targeted synthetic JAKis suggest differential pain modification to bDMARDs,27 implying the presence of distinct biological drivers of pain that are independent to the peripheral sensitisation of synovitis. Such data raise interesting hypotheses on the role of JAKi in pain pathways and highlight the importance of not restricting evaluation of refractory disease to just that associated with synovial inflammation.
Concluding remarks
This viewpoint highlights the knowledge gap in the identification and understanding of refractory RA disease. In the absence of well-phenotyped studies and a systematic approach to evaluating refractory RA disease, the true extent, impact and underlying basis remains unclear. Disease duration and damage that are associated with a suboptimal patient response profile blur the precision with which we may be sequencing therapies and estimating the size of the problem. Suboptimal targeting of disease from the outset may also be implicated in the development of refractory pathology. The heterogeneous nature of RA and pharmacokinetic drivers of drug response both likely play a role in leading to this clinical landscape. Continued drug development pipeline, therefore, remains important to offer ongoing opportunities.
Acknowledgments
The author acknowledges and thanks Professor Dennis McGonagle for his critical review of the article.
References
Footnotes
Handling editor Josef S Smolen
Contributors MHB was solely responsible for initiating and writing the submitted manuscript.
Funding MHB is partly supported by the NIHR Leeds Biomedical Research Centre.
Competing interests None declared.
Patient consent Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Correction notice This article has been corrected since it published Online First. Figures 1 and 2 have been corrected.