Article Text

Abstract
Objectives Patients with rheumatoid arthritis and spondyloarthritisshow higher mortality rates, mainly caused by cardiac comorbidities. The TghuTNF (Tg197) arthritis model develops tumour necrosis factor (TNF)-driven and mesenchymalsynovial fibroblast (SF)-dependent polyarthritis. Here, we investigate whether this model develops, similarly to human patients, comorbid heart pathology and explore cellular and molecular mechanisms linking arthritis to cardiac comorbidities.
Methods Histopathological analysis and echocardiographic evaluation of cardiac function were performed in the Tg197 model. Valve interstitial cells (VICs) were targeted by mice carrying the ColVI-Cretransgene. Tg197 ColVI-Cre Tnfr1 fl/fl and Tg197 ColVI-Cre Tnfr1 cneo/cneo mutant mice were used to explore the role of mesenchymal TNF signalling in the development of heart valve disease. Pathogenic VICs and SFs were further analysed by comparative RNA-sequencing analysis.
Results Tg197 mice develop left-sided heart valve disease, characterised by valvular fibrosis with minimal signs of inflammation. Thickened valve areas consist almost entirely of hyperproliferative ColVI-expressing mesenchymal VICs. Development of pathology results in valve stenosis and left ventricular dysfunction, accompanied by arrhythmic episodes and, occasionally, valvular regurgitation. TNF dependency of the pathology was indicated by disease modulation following pharmacological inhibition or mesenchymal-specific genetic ablation or activation of TNF/TNFR1 signalling. Tg197-derived VICs exhibited an activated phenotype ex vivo, reminiscent of the activated pathogenic phenotype of Tg197-derived SFs. Significant functional similarities between SFs and VICs were revealed by RNA-seq analysis, demonstrating common cellular mechanisms underlying TNF-mediated arthritides and cardiac comorbidities.
Conclusions Comorbidheart valve disease and chronic polyarthritis are efficiently modelled in the Tg197 arthritis model and share common TNF/TNFR1-mediated, mesenchymal cell-specific aetiopathogenic mechanisms.
- tnf-alpha
- cardiovascular disease
- fibroblasts
- rheumatoid arthritis
- spondyloarthritis
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Chronic inflammatory joint diseases are associated with articular inflammation leading to joint damage and increased mortality rates, which are mainly attributed to cardiovascular comorbidities.1 2 Cardiac disease manifestations are detected in 70%–80% of patients with rheumatoid arthritis (RA) and spondyloarthritis (SpA) and symptoms can vary greatly, including arrhythmias, ischaemic heart failure as well as valvular diseases, such as valve insufficiency and stenosis.3 4 The mechanisms mediating the co-occurrence of cardiac comorbidities in patients with chronic inflammatory joint diseases remain unknown.
The critical role of tumour necrosis factor (TNF) in RA and SpA pathologies is now well established both in transgenic animal models5–8 and by the highly positive clinical responses of human patients to anti-TNF therapies.9 10 Interestingly, recent studies in mice with deregulated TNF expression indicated a pivotal role of TNF also in cardiovascular diseases.11–15 It, therefore, appears that TNF may commonly underlie arthritis and arthritis-related cardiac manifestations in human patients, which could also explain the amelioration of both of these comorbidities in patients treated with anti-TNF biologics.16
Mesenchymal cells are active participants in the structure and function of almost all tissues and contribute to their homeostasis.17 The mesenchymal cells of the joint are the synovial fibroblasts (SFs).18 19 Several studies have implicated SFs as key pathogenic cells, capable of initiating and driving the development of joint pathologies both in mouse models and human patients.8 20 21 Interestingly, using TNF-driven models of comorbid arthritis and inflammatory bowel disease (IBD), we have previously established that TNF signals, uniquely operating in SFs or intestinal mesenchymal cells (IMCs), are sufficient to orchestrate the full pathogenic process of these two complex pathologies.8 Yet another mesenchymal cell type, which is known to form the heart valves and is responsible for the maintenance of valve extracellular matrix structures, is the valve interstitial cells (VICs).22–24 Recent in vitro studies have suggested that TNF can activate quiescent VICs into myofibroblasts inducing their pathogenic contribution to heart valve diseases (HVDs).23 25 We have therefore hypothesised that the huTNF-driven, Tg197 model of arthritis5 may exhibit heart valve pathology and that a common mesenchymal cell-specific TNF-mediated mechanism, operating on VICs, could explain the comorbidity.
We show here that Tg197 mice develop spontaneous left-sided heart valve pathology, characterised by extensive fibrosis and thickening of the aortic valve (AV) and mitral valve (MV) and associated with activated and hyperproliferating VICs. Valvular stenosis was associated with deterioration of cardiac function due to valvular degeneration and left ventricular (LV) dysfunction, simulating comorbid valvular diseases detected in patients with RA/SpA. Moreover, we show that this cardiac phenotype is ameliorated on Ab-mediated inhibition of TNF or by genetic mesenchymal-specific ablation of TNFR1. We further demonstrate that Tg197 VICs cultured ex vivo exhibit an activated phenotype characterised by increased huTNF production as well as increased proliferative and migratory capacities, similar to the one exhibited by arthritogenic SFs. Comparison of RNA-sequencing profiles between Tg197-derived SFs and VICs revealed similar pathogenic genes and pathways being activated in the two cell types. Overall, our studies establish a common TNF-driven mesenchymal cell-specific mechanism that may underlie aetiopathogenesis of comorbid joint and HVDs also in human patients.
Materials and methods
Mice
Tg197,5 ColVI-Cre,8 Tnfr1 fl/fl 26 and Tnfr1cneo/cneo 27 mice were previously described; Rosa26 mT/mG mice28 were purchased from the Jackson Laboratories. Mice were maintained on a C57BL/6J or C57BL/6×CBA genetic background in the animal facilities of Biomedical Sciences Research Center (BSRC) ‘Alexander Fleming’ under SPF conditions. All animals were sacrificed at 11–12 weeks of age. Further details can be seen online in supplementary methods.
Supplementary file 1
Antibodies
Antibodies used for immunohistochemistry, fluorescence-activated cell sorting (FACS) and immunofluorescence can be found in online supplementary methods.
Immunohistochemistry and immunofluorescence
Paraffin-embedded tissue sections and heart transverse optimal cutting temperature (OCT) cryosections were stained and evaluated according to protocols found in online supplementary methods.
Echocardiography and ECG
Echocardiography assessment and ECG were performed in the Department of Pharmacology, Medical School NKUA, Greece. Further details can be seen in online supplementary methods.
Isolation and culturing of SFs and VICs
SFs and VICs were isolated and cultured up to the third or fourth passage when they were used for cellular assays and sequencing. Detailed protocol is described in online supplementary methods.
FACS
See details in online supplementary methods.
Proliferation assay
To determine cellular proliferation, the Cell Proliferation ELISA, BrdU kit (Sigma-Aldrich) was used.
ELISA
Detection of hTNF was performed using the hTNF Quantikine Elisa (R&D Systems).
Wound-healing assay
To determine the migratory capacity of the cells, we used the wound-healing assay as described in online supplementary methods.
3′ RNA-sequencing and analysis
RNA-seq was performed in three biological replicates of cultured VICs and SFs isolated from Tg197 mice and WT littermates at their eighth week of age. Further analysis is found in online supplementary methods.
Statistical analysis
Data are presented as mean±SEM, and Student’s t test was used for the evaluation of statistical significance, with P values <0.05 being considered statistically significant. Analysis was performed using the GraphPad Prism V.6.
Results
TNF-dependent left-sided heart valve pathology develops as a comorbid condition in the Tg197 arthritis model
Histopathological evaluation of heart tissue from Tg197 animals revealed pathological alterations localised in the left side of the heart, affecting specifically the AV (figure 1A) and MV area (figure 1B), while the pulmonary valve as well as the blood arteries and vessels appeared unaffected (online supplementary figure S1,4). Pathology was associated with AV and MV thickening (figure 1C) mainly due to fibrosis, which extended to the root of the valve, as shown by the intense Masson’s staining (figure 1A,B). Inflammation appeared to have only a minimal contribution, as indicated by the limited number of infiltrating inflammatory cells in the valves at 12 weeks of age (online supplementary figure S3).

Tg197 arthritis model develops tumour necrosis factor (TNF)-dependent left-sided heart valve disease which leads to left ventricle (LV) dysfunction. (A, B) Representative images of H&E and Masson’s trichrome-stained transverse heart sections showing the aortic valve (AV) (A) and the mitral valve (MV) (B) leaflets of Tg197 and WT littermate animals at 12 weeks old of age;(scale bar, 400 μm) (C) Comparison of the AoV and MV thickness between WT, Tg197 and Tg197 treated with anti-TNF infliximab (Remicade) animals at 11–12 weeks of age (data are presented as individual values, with mean±SEM; *P<0.02; **P<0.01). (D) Blood aortic (AoV) and mitral velocities (MV E and A) acquired by Doppler analysis of Tg197 mice and WT littermates at their 12 weeks of age (left and right panels, respectively; data are presented as individual values, with mean±SEM; ***P<0.0001). (E) Ejection fraction (EF%) of Tg197 mice and WT littermates at their 12 weeks of age, calculated by the modified Simpson equation, using 2D images in echocardiography analysis (data are presented as individual values, with mean±SEM; ***P<0.0001). (F) Representative ECGs of Tg197 animals with few minutes interval (four consecutive time points with ~1 min interval, starting from the upper panel), at their 12 weeks of age. AVR, aortic valve root; WT, wild type.
Signs of heart valve pathology were detected in the Tg197 mice already from 4 weeks of age and became progressively worse as animals aged (online supplementary figure S4) in parallel to their arthritis pathology. By 8 weeks of age, when Tg197 mice had established arthritis, pathology in both valves was manifested with 100% penetrance and without a gender bias. Treatment of Tg197 animals with the anti-TNF infliximab (Remicade), from 4 to 11 weeks of age, resulted in the amelioration of the HVD, demonstrated by the decrease in valvular thickening and fibrosis (figure 1C and supplementary figure S2)
Heart valve pathology leads to LV dysfunction in the Tg197 animals
To assess whether the valvular thickening and fibrosis observed in Tg197 animals also affect their cardiac function, we performed echocardiography and ECG analysis in 12-week-old mice. Tg197 mice displayed increased AV and MV velocities (figure 1D), indicative of valvular stenosis. Moreover, in approximately 15% of the transgenic mice examined, aortic and/or mitral regurgitation was detected (online supplementary figure S5), suggesting valvular insufficiency. An additional consequence of the MV dysfunction was the observed increased atrial pressure leading to dilation of the left atrium (LA) (table 1). Echocardiography data analysis consistently showed that Tg197 animals displayed LV dilation with some degree of hypertrophy, indicated by the increased LV dimensions (LV end-diastolic diameter (LVEDd), LV end-systolic diameter (LVEDs), LV length in diastole (LVLd), LV end-diastolic posterior wall thickness (LVPWd), end-diastolic interventricular septal thickness (IVSd)) and the significant increase of heart-to-body weight ratio compared to WT mice (table 1). Tg197 mice also exhibited significant reduction of the global cardiac function, as indicated by their reduced ejection fraction (EF%) (figure 1E) and, more importantly, by their reduced regional contractile function reflected in the lower systolic velocity of the posterior wall (SVPW) (table 1).
Echocardiographic parameters in Tg197 and WT mice at 12 weeks of age
Interestingly, we have observed that Tg197 animals exhibit increased premature mortality of unknown aetiology starting at 10 weeks of age, reaching an ~50% incidence at 13 weeks of age (online supplementary figure S6). Notably, assessment of cardiac function of Tg197 animals showed that they were prone to exhibit fatal episodes of arrhythmias in advanced disease stages (12 weeks), mainly switching from bradycardia (~200 bpm) to tachycardia (~500–650 bpm) in a few minutes interval during ECG (figure 1F). Therefore, arrhythmic episodes could be associated with the premature deaths observed in Tg197 animals.
Collectively, our data show that the histopathological findings in Tg197 heart valves are associated with left-sided valvular degeneration and dysfunction and are accompanied by echocardiographic findings of LV cardiomyopathy.
Hypertrophic valves of Tg197 mice consist mainly of activated VICs
Since SFs have been previously established as drivers of arthritogenesis in the Tg197 model,8 we investigated whether VICs play also a pathogenic role in the observed Tg197 heart valve pathology. To this end, we first crossed the reporter mouse Rosa26 mT/mG which expresses green fluorescent protein (GFP) upon recombination, with the ColVI-Cre mouse, which has been previously used to target mesenchymal cells in the joints, small intestine,8 colon29 and other organs.30 Examination of the heart valves of ColVI-Cre-Rosa26 mT/mG mice revealed co-localisation of GFP expression with vimentin (figure 2A), a known marker of fibroblasts and VICs,23 31 indicating efficient targeting of VICs by ColVI-Cre, and confirming their mesenchymal origin.22 Efficient recombination was confirmed by further characterisation of GFP+ cells derived from dissociated heart valve tissue from ColVI-Cre-Rosa26 mT/mG mice using FACS analysis. GFP+ cells strongly expressed VIC and mesenchymal cell markers (vimentin, CD29 and podoplanin), while displaying no expression of haematopoietic (CD45) and endothelial (CD31) markers (figure 2B). These results suggest that the ColVI-Cre mouse effectively targets mainly vimentin+, CD29+ and podoplanin+ mesenchymal-like VICs in the heart valve.

Heart valve disease of Tg197 mice is caused by accumulation of activated mesenchymal valve interstitial cells (VICs). (A) Representative images of transverse heart cryosections of ColVI-Cre-Rosa26 mT/mG mice, at their 8 weeks of age, and colocalisation of GFP expression with vimentin expression in the heart valve (lower panel: higher magnification of the upper panel) (scale bar: 100 μm). (B) Fluorescence-activated cell sorting (FACS) analysis of ColVI-expressing cells (GFP+) with markers for endothelial (CD31), haematopoietic (CD45) and fibroblast/mesenchymal cells (CD29, podoplanin [PDPN], vimentin) from dissociated heart valves of ColVI-Cre-Rosa26 mT/mG mice, at their 8 weeks of age (data are presented as mean±SEM, n=3 from three individual experiments). (C) Representative images of transverse heart cryosections of ColVI-Cre mT/mG and Tg197 ColVI-Cre mT/mG mice at their 12 weeks of age (scale bar: 100 μm). (D) Representative image of transverse heart cryosections of Tg197 mice at their 12 weeks of age and colocalisation of smooth muscle actin (α-SMA) expression with vimentin expression in the heart valve and root (scale bar: 100 μm).
To explore the role of VICs in the Tg197 heart valve pathology, we crossed the ColVI-Cre-Rosa26 mT/mG mice with Tg197 mice. The thickened fibrotic heart valves of these mice were mainly populated by GFP+ VICs (figure 2C), supporting their central role in the heart valve phenotype. The pathogenic potential of VICs in Tg197 animals was further assessed by the expression of α-SMA, a well-established marker of activated myofibroblastic VICs.32 Interestingly, α-SMA-expressing VICs were detected in the thickened valvular area and root of Tg197 mice (figure 2D), indicating that the pathology observed is mainly characterised by accumulation of activated VICs.
TNFR1 signalling in mesenchymal cells is necessary and sufficient for the development of Tg197 heart valve pathology
Having established the contribution of activated mesenchymal VICs and TNF dependency of the valvular hyperplasia in Tg197 mice, we further explored the role of mesenchyme-specific TNF signalling in the development of this pathology. To address whether TNF signalling in mesenchymal VICs is required for the development of heart valve pathology, Tg197 animals were crossed with ColVI-Cre Tnfr1 fl/fl animals.26 Tg197 ColVI-Cre Tnfr1 fl/fl mice exhibited ameliorated heart valve pathology, as indicated by the lack of heart valve thickening and fibrosis (figure 3A, C). This finding suggests that TNF signalling, through TNFR1 in mesenchymal cells, is essential for the pathogenesis of HVD in the Tg197 model.
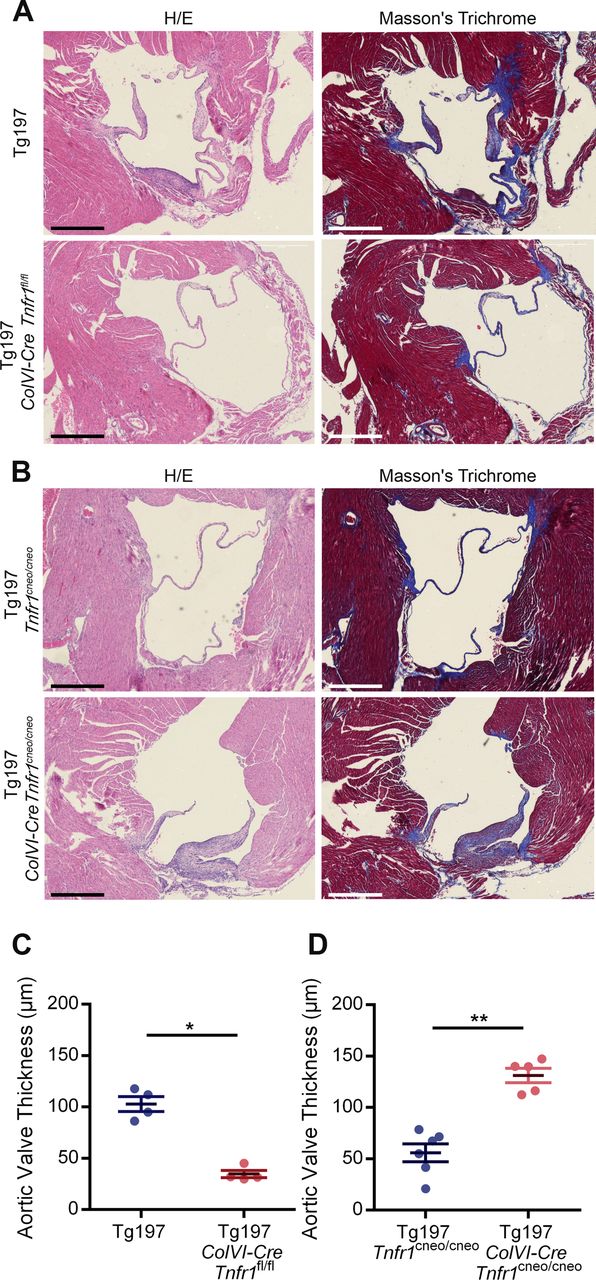
TNF signalling on valve interstitial cells (VICs) is required and sufficient for the development of heart valve disease of Tg197 animals. (A) Representative images of H&E and Masson’s trichrome-stained transverse heart sections of Tg197 and Tg197 ColVI-Cre Tnfr1 fl/fl and animals at 12 weeks of age (scale bar, 500 μm). (B) Representative images of H&E and Masson’s trichrome-stained transverse heart sections of Tg197 Tnfr1 cneo/cneo and Tg197 ColVI-Cre Tnfr1 cneo/cneo and at 12 weeks of age (scale bar, 500 μm). (C) Comparison of the aortic valve thickness between Tg197 and Tg197 ColVI-Cre Tnfr1 fl/fl animals at 12 weeks of age (data are presented as individual values, with mean± SEM; *P<0.03). (D) Comparison of the aortic valve thickness between Tg197 Tnfr1 cneo/cneo and Tg197 Tnfr1 cneo/cneo at 12 weeks of age (data are presented as individual values, with mean± SEM; **P<0.005).
Next, we examined whether TNF signalling in mesenchymal cells was also sufficient to induce heart valve pathology in Tg197 mice. To this end, we crossed Tg197 with ColVI-Cre Tnfr1 cneo/cneo mice to achieve specific reactivation of TNFR1 signalling only in mesenchymal cells.27 Tg197 ColVI-Cre Tnfr1 cneo/cneo mice developed valvular thickening and extensive fibrosis, while control Tg197 Tnfr1 cneo/cneo did not show any signs of heart valve thickening and fibrosis (figure 3B,D), demonstrating that TNF signalling through TNFR1 in mesenchymal cells is sufficient to trigger heart valve pathology in Tg197 mice. Consequently, TNF signalling in the mesenchyme is both necessary and sufficient for the development of heart valve pathology in Tg197 animals.
Ex vivo-derived Tg197 VICs exhibit an activated phenotype
It is known that ex vivo human RA and mouse arthritogenic SFs exhibit increased proliferative and migratory capacities.33–35 To investigate whether pathogenic VICs display a similar phenotype, we isolated VICs from Tg197 and WT animals at 8 weeks of age, when HVD is well established.
We first confirmed the homogeneity of VICs cultures by characterising cultured VICs isolated from ColVI-Cre-Rosa26 mT/mG animals. FACS analysis confirmed that approximately 80% of the isolated VICs were GFP+ and displayed high expression of known fibroblast and mesenchymal cell markers including CD29, vimentin, podoplanin, CD140a, CD90.2, CD105, vascular cell adhesion protein 1 (VCAM-1) and intercellular adhesion molecular 1 (ICAM-1) (figure 4A), while they lacked expression of haematopoietic (CD45) and endothelial (CD31) markers, thus preserving the observed in vivo expression marker profile (figure 2B).
We further assessed the activation status of Tg197-derived VICs. These cells were found to overexpress hTNF (figure 4B) and displayed increased proliferative and migratory capacities (figure 4C,D), similarly to Tg197-derived SFs.34 Therefore, VICs are shown to exhibit an activated phenotype with similar characteristics to the one exhibited by the arthritogenic Tg197 SFs ex vivo.33 34

Activated phenotype of Tg197-derived valve interstitial cells (VICs) ex vivo. (A) Fluorescence-activated cell sorting (FACS) analysis of ColVI-expressing cells (GFP+) with markers for endothelial (CD31), haematopoietic (CD45) and fibroblast/mesenchymal cells (CD29, podoplanin [PDPN], vimentin, CD140a, CD90.2, CD105, VCAM-1, ICAM-1) in isolated VICs from ColVI-Cre-Rosa26 mT/mG mice at 8 weeks of age. (B–D) Levels of secreted hTNF in the supernatants (B), BrdU incorporation (C) and wound healing ability calculated by percentage of wound closure (D) of VICs isolated from WT and Tg197 mice at 8 weeks of age (data are presented as mean±SEM, n=3 from three individual experiments; **P<0.001; ***P<0.0005).
Tg197 VICs express common pathogenic signatures with Tg197 SFs
Arthritogenic SFs have been recently found to exhibit a distinct expression profile, characterised by pathogenic deregulation of genes affecting key pathways for the development of polyarthritis symptoms.36 We, therefore, explored the commonalities of pathogenic Tg197 VICs and SFs at the gene expression, pathway and transcriptional regulation level. For this purpose, we isolated SFs and VICs from 8-week-old Tg197 animals, with established arthritis and HVD, and compared their expression profiles to those of SFs and VICs isolated from WT littermates by using RNA-sequencing.
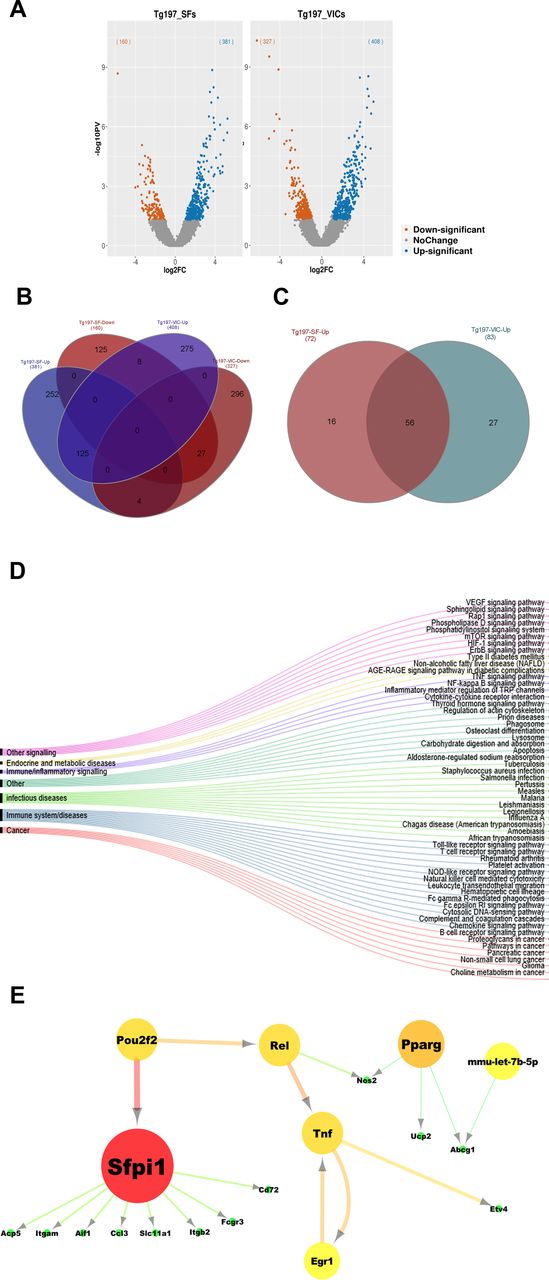
Both Tg197 SFs and VICs displayed >500 significant differentially expressed genes (DEGs) compared to their WT controls (figure 5A). More specifically, a total of 408 and 381 genes were upregulated in Tg197 VICs and SFs, respectively, with almost 30% of them commonly upregulated in both cell types (figure 5B), while a total of 327 and 160 genes were downregulated in Tg197 VICs and SFs, respectively, with approximately 10% of them commonly downregulated in both cell types (figure 5B). Further functional enrichment analysis of the common upregulated genes placed immune and inflammatory responses, as well as nuclear factor (NF)-κB signalling at the top enriched pathways. Pathways enriched in the overlapping downregulated genes included extracellular matrix organisation and regulation of growth, indicating extracellular matrix (ECM) remodelling and deregulated cell growth (online supplementary figure S7).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Tg197 valve interstitial cells (VICs) exhibit common pathogenic molecular signatures with Tg197 synovial fibroblasts (SFs) at 8 weeks of age. (A) Volcano plots with the number of differentially expressed genes (DEGs) of Tg197 VICs and SFs compared to WT. (B) Venn diagram showing the overlap of DEGs in Tg197 VICs and SFs. (C) Venn diagram showing overlap of enriched KEGG pathways derived from functional enrichment analysis of upregulated DEGs in Tg1917 VICs and SFs. (D) Alluvial diagram illustrating overlapping KEGG pathways in Tg197 VICs and SFs, grouped according to their KEGG broader categories. (E) Intersection of regulatory networks of Tg197 VICs and SFs.
To further explore the similarities of these two pathogenic cell types at the pathway level, functional enrichment analysis was performed for all DEGs in SFs and VICs. Interestingly, KEGG pathways enriched in SFs’ and VICs’ upregulated genes show a great overlap (60%) (figure 5C). These pathways were subsequently clustered into broader KEGG pathway categories. The most pronounced category was immune response, which included pathways such as chemokine and TLR signalling, while the most prominent correlation was to human ‘rheumatoid arthritis’ term, with known RA-related and cardiovascular disease-related genes (Tnf, Il1b 37 and Acp5 38) being upregulated in both cell types. Other categories include cancer and infectious diseases, such as tuberculosis and pertussis which have also been associated with inflammation and TNF signalling. TNF and NF-κB signalling were also enriched in both cell types, with a distinct set of genes such as Mmp9, Tnf, Il1b, Cxcl2;3;12, Ccl4 and Cd14 being upregulated in both Tg197 SFs and VICs (figure 5D). Interestingly, some of the functions enriched only in VICs’ DEGs involve cardiovascular diseases, indicating the differences between VICs and SFs due to their different tissue of origin (online supplementary figure S7).
Furthermore, the RNEA tool,39 which infers regulatory networks by predicting interactions between transcription factors and their target genes, was used to explore the similarities between VICs and SFs at the transcriptional regulation level. Regulatory networks were extracted from both cells’ gene expression profile and their intersection is reported in figure 5E. Interestingly, Sfpi1 and Pparg, which are known to regulate mesenchymal activation,36 were revealed by this analysis as the two main common regulators of the two networks; Tnf was also found to be a central regulator. These findings strongly suggest that Tg197 VICs share a commonly altered expression profile with Tg197 SFs at the gene expression, functional pathways and transcriptional regulation circuit levels.
Discussion
Patients with RA and SpA show a higher risk of developing associated cardiac diseases, which highly contribute to their increased mortality rates.2 More specifically, they exhibit a 30% increased incidence of valvular pathologies, including non-specific valvular thickening and mild valve regurgitation.3 40 Recent studies using sensitive imaging methods, such as transesophageal echocardiography, report an even greater prevalence of left-sided HVD in RA patients with valve thickening in half of the cases involving both mitral (47%) and aortic valves (32%) and valve regurgitation (21%).41 The involvement of TNF in the pathogenesis of RA and SpA is well established; however, its role in the development of arthritis-related cardiac comorbidities remains unknown.
We demonstrate here that overexpression of TNF, in the TghuTNF (Tg197) arthritis model, in addition to the chronic polyarthritis5 drives also the development of spontaneous left-sided HVD, which mainly leads to valvular thickening with some degree of stenosis and occasionally to valve insufficiency, comorbid pathologies often observed in patients with RA/SpA.3 42 Interestingly, a similar left-sided heart valve pathology, exhibiting valvular thickening and fibrosis, was also observed in the TgA86, transmembrane TNF overexpressing, mouse model of SpA6 43 (supplementary figure S8), further strengthening the pathogenic role of TNF in the development of arthritis-related cardiac comorbidities. The greater mechanical stress and haemodynamic pressures imposed on the left side of the heart is a likely explanation for the discrepancy between the diseased left-sided and unaffected right-sided valves, also observed in patients with RA/SpA.
AV stenosis and MV and/or AV regurgitation have been shown to result in LV hypertrophy with preserved EF and occasionally in LV dilation with some degree of contractile dysfunction.44 Similarly, in the Tg197 model, valvular pathology contributes to the observed extensive LV dilation with some degree of LV hypertrophy as well as to the concomitant contractile dysfunction. However, additional mechanisms that have been proposed as contributing factors in the increased prevalence of global heart failure in patients with RA/SpA, such as myocardial fibrosis and oedema as well as arterial blood pressure, coronary heart disease and myocardial remodeling45 remain to be studied for their contribution in the global heart impairment observed in Tg197 animals. We have also detected repeated arrhythmic episodes in Tg197 mice which could explain the premature sudden deaths observed in this model recapitulating the increased risk of sudden cardiac death experienced by patients with RA/SpA, due to atrial fibrillation and other types of tachyarrhythmias which suggest diffuse myocardial electrical instability.46 47 Overall, our data demonstrate that the Tg197 arthritis model develops HVD and cardiac arrhythmias that closely mimic the cardiac clinical findings and premature mortality observed in patients with arthritis, supporting the value of this model in providing mechanistic insights into the pathogenesis of these comorbidities. The reversal of the cardiac phenotype by pharmacological inhibition of TNF in this model supports the vital role of TNF in the development of RA/SpA-related cardiac valvular comorbidities and suggests that anti-TNF therapy could also prevent cardiac comorbidities and avoid adverse cardiovascular side effects caused by other drugs, such as DMARDs.2 Our findings also highlight the importance of regular echocardiographic screening on patients with RA and SpA.
The association between elevated TNF levels and valvular pathology has been previously suggested in other mutant mice.14 15 Notably, these mice exhibit an inflammatory valvulitis, in contrast to the hypertrophic valves of Tg197 mice, which consist mainly of activated mesenchymal VICs. This discrepancy could be attributed to various factors, such as differences in the genetic background or in the cytokine imbalances driving the pathology. More specifically, the inflammatory phenotype of IL1Ra-deficient mice14 has been observed in the inflammation-susceptible48 BALB/c genetic background whereas in the C57 background they show milder pathology.14 Additionally, pathology in the IL1Ra-14 as well as in the TTP-deficient15 and K/BxN transgenic49 mice could be driven by diverse upstream mechanisms providing additional pathogenic cytokine disbalances apart from TNF.
Extensive characterisation and comparison of the transcriptional profiles of pathogenic Tg197 VICs and SFs, compared with their healthy counterparts, revealed a shared altered and pathogenic profile of these two cell types. Inflammatory and immune responses were among the commonly enriched KEGG pathways in both Tg197 SFs and VICs, supporting their activated and pathogenic status. Our analysis further supports the central role of Tnf in both cell types and pathologies. Interestingly, Sfpi1, an NF-κB modulator,50 emerged as a common transcriptional regulator of both activated VICs and SFs, highlighting the importance of NF-κB signalling in this process, as was also confirmed by the enrichment of the NF-κB signalling pathway in both cell types. Moreover, Sfpi1, encoding the myeloid-specific transcription factor PU.1,51 has been found to be upregulated in RA-FLS,52 while being also implicated in the pathogenesis of heart hypertrophy.53 Collectively, the centrality of Sfpi1, in combination with the enriched immune and TLR signalling, as well as NF-κB signalling pathways, in both Tg197 VICs and SFs could support their conversion to activated mesenchymal cells possessing pathogenic innate immune properties. This hypothesis is further supported by recent findings suggesting that Tg197 SFs undergo a metabolic reprogramming,54 similar to the metabolic alterations reported in both inflammatory heart diseases and RA.55 56 Therefore, we hypothesise that VICs and SFs become pathogenic on common TNF-induced metabolic reprogramming acquiring a detrimental innate phenotype, which should be further explored.
We finally show here that mesenchymal-specific TNF signalling, through TNFR1, is both required and sufficient for the development of heart valve pathology. Notably, SF-specific and IMC-specific TNFR1 signalling has been previously demonstrated to be causal in orchestrating comorbid polyarthritis and Crohn’s-like IBD in a TNF overexpressing mouse model.8 It may, therefore, be strongly postulated that mesenchymal cell responses to TNF explain complex chronic inflammatory disease comorbidities involving joint, intestinal and, as shown in the present study, also cardiac pathologies. Future detailed insights into the molecular and cellular mechanisms commonly underlying aetiopathogenesis of mesenchymal cell-driven comorbidities, such as those expressed under the RA/SpA paradigm, may also offer novel approaches to therapeutically target common pathogenic processes.
Acknowledgments
The authors are grateful to Dr Branko Simic, Professor Thomas F Lüscher and Dr Stelios Psarras for their scientific input and advice, as well as Dr Vaggelis Harokopos, Dr Panagiotis Moulos and the personnel of BSRC “Alexander Fleming” Genomics Facility for performing RNA-sequencing and and raw data analysis, Anna Katevaini and Christina Geka for histology, Michalis Meletiou for technical assistance and Ana Henriques for help in figure preparation.
References
Footnotes
LN and MS contributed equally.
Handling editor Josef S Smolen
Contributors GK, LN, MS, NK and MCD designed the study and interpreted the experimental results. LN, MS, PC and IM performed the experiments and data analysis. AP contributed to the data analysis. LN and MCD wrote the first draft of the manuscript and all authors were involved in critically revising its final preparation. All authors approved the final version to be published.
Funding This work has been funded by the IMI project BTCure (GA no. 115142-2), the Advanced ERC grant MCs-inTEST (340217), and a ’Research Project for Excellence IKY/Siemens' (GA no. 3288) to GK, an ’IKY fellowship of Excellence for postgraduate studies in Greece – Siemens program' to MS, the Operational Program “Competitiveness, Entrepreneurship and Innovation 2014-2020”, co-financed by Greece and the European Union (European Regional Development Fund) (MIS 5002562-KRIPIS II) as well the grant GA 31354 (CodeAge) FP7 -PEOPLE-2011-ITN to LN. The authors also wish to thank the InfrafrontierGR Infrastructure (co-funded by the ERDF and Greek NSRF 2007-2013) for providing mouse hosting and phenotyping facilities, including histopathology, flow cytometry and advanced microscopy.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.