Article Text

Abstract
Background Targeted inhibitors of B-cell activating factor (BAFF) have been evaluated in phase III trials in over 4000 patients with systemic lupus erythematosus (SLE). Post hoc analyses of these studies identify greater treatment effect in patients entering with higher disease activity, greater corticosteroid doses, anti double-stranded DNA (dsDNA) and low complement C3 or C4.
Objectives To evaluate the efficacy and safety of blisibimod, a BAFF inhibitor, in a population of patients with SLE enriched for high disease activity.
Methods 442 patients with SLE with antinuclear antibodies or anti-dsDNA and Safety of Estrogen in Lupus Erythematosus National Assessment – Systemic Lupus Erythematosus Disease Activity Index (SELENA-SLEDAI) score ≥10 on standard-of-care medications were randomised to receive weekly subcutaneous blisibimod (200 mg) or placebo. Corticosteroid taper was encouraged from week 8. The primary end point was the week 52 SLE Responder Index-6 (SRI-6).
Results The SRI-6 primary end point was not met. There was a statistically significant steroid-sparing effect, and significantly more blisibimod-treated subjects achieved corticosteroid taper. Increased blisibimod treatment effect on SRI-6 was observed in subjects who achieved a concomitant decrease in corticosteroid dose from baseline. In subjects with baseline urinary protein:creatinine ratio (UPCR) ≥56.5 mg/mmol, significantly higher proportions of blisibimod subjects achieved >50% reduction in UPCR and/or UPCR <56.5 mg/mmol. Reductions in SLE autoantibodies and B cells, and increases in complement C3 and C4 were observed with blisibimod.
Blisibimod was well-tolerated. The most common adverse events were upper respiratory tract infection, urinary tract infection, injection site erythema/reaction and diarrhoea.
Conclusions Although the SRI-6 end point was not met, blisibimod was associated with successful steroid reduction, decreased proteinuria and biomarker responses.
Trial registration number NCT01395745.
- B cells
- systemic lupus erythematosus
- corticosteroids
Statistics from Altmetric.com
Introduction
Targeted, biologic inhibitors of B-cell activating factor (BAFF), belimumab and tabalumab, have been evaluated in four large phase III clinical trials in nearly 4000 patients with systemic lupus erythematosus (SLE).1–4 Data from these studies indicate that the greatest treatment effect is discernible using more stringent end points and focusing on patients who enter with higher disease activity scores, higher corticosteroid doses, demonstrable antidouble-stranded DNA (dsDNA) and low complement C3 or C4.5 6
These observations provide important guidance to clinical trial design, especially in light of the too-common failures of large clinical trials in SLE. Moreover, the data provide important guidance to treatment practice where use of high-dose corticosteroids, immunosuppressive agents and cytotoxic agents remain the standard-of-care for moderate and severe SLE despite their toxicities.7 8
Blisibimod (A-623, AMG 623) is a potent and selective BAFF inhibitor composed of a tetrameric BAFF binding domain fused to a human IgG1 Fc region.9 Although the phase II Placebo-controlled Evaluation of A‑623 Response in Lupus (PEARL-SC) trial with blisibimod in SLE failed to meet the primary efficacy end point, SLE Responder Index-5 (SRI-5), it corroborated the observations from larger Phase III trials in that better treatment effect was observed in subjects with higher disease activity, taking corticosteroids, who had abnormalities in anti-dsDNA and/or complement.10 Furthermore, in keeping with the belimumab observations,11 favourable effects on proteinuria were observed.10 In addition, blisibimod treatment was associated with significant decreases in anti-dsDNA, immunoglobulins, total B-cell counts and fatigue12 and significant increases in complement C3 and C4. Accordingly, the phase III Clinical and Health Assessments with BLISibimod SC, Study 1 (CHABLIS-SC1) trial prospectively evaluated a population of subjects with SLE enriched for the traits that, in earlier studies, were associated with greater treatment effect relative to background standard-of-care treatments.
Methods
Patient population and trial design
The CHABLIS-SC1 study (NCT01395745) was conducted in Belarus, Brazil, Colombia, Georgia, Guatemala, Hong Kong, Korea, Singapore, Malaysia, Mexico, Russia, Sri Lanka, Taiwan, Thailand and the Philippines. The study enrolled 442 adults aged ≥18 years who were seropositive for anti-nuclear antibody (ANA) (≥1:80 in immunofluorescence assay) or anti-dsDNA (≥30 IU/mL), met at least four of the SLE Classification Criteria defined by the American College of Rheumatology (ACR),13 and had a baseline disease activity score of ≥10 on the SELENA-SLEDAI scale despite concomitant and stable use of systemic corticosteroids at the lesser of 0.5 mg/kg or 40 mg/day of prednisone or equivalent. Other standard-of-care medications in stable doses were allowed (antimalarials, methotrexate, mycophenalate, azathioprine, leflunomide, non-steroidal anti-inflammatory drugs). Subjects were excluded from study if they had active vasculitis, central nervous system lupus or severe active lupus nephritis, proteinuria >6 g/24 hours (or equivalent) or a history of malignancy in the last 5 years, HIV infection, hepatitis B or C infection or tuberculosis or recent use of a B-cell targeted drug, investigational agents, cyclophosphamide or other alkylators, transfusion, intravenous immunoglobulin, plasmapheresis, plasma exchange, high-dose corticosteroid, antitumour necrosis factor-α or other biologics, ciclosporin or live vaccines.
Eligible subjects were randomly assigned using an interactive web-based randomisation system in a 5:4 ratio to receive subcutaneous blisibimod 200 mg once weekly or corresponding volume and frequency-matched placebo using a double-blind interactive web response system. The dose of blisibimod in this trial was based on the observations from a prior Phase II trial in SLE.10 Randomisation was stratified by race (African vs non-African race), baseline SELENA-SLEDAI score (≤12 vs >12) and proteinuria (<2 vs ≥2 g/24 hours equivalent). Screening was initiated in March 2013. The sponsor, all investigators, study personnel and subjects remained blind to treatment allocation until after the effects on SRI-6 at week 52 were analysed in November 2016.
Subjects were required to continue background SLE medication at the stable, prestudy doses throughout the study except for corticosteroids. Corticosteroids could be increased during the first 8 weeks of the trial but were not allowed not to exceed 5 mg or 25% above baseline (whichever was lower) by week 8, after which time a subject at higher dose would be considered a treatment failure. Steroid taper was encouraged from week 8 onwards with a goal of achieving ≤7.5 mg prednisone/day or equivalent. Subjects received study drug for 53 weeks.
The study was conducted in accordance with the provisions of the Declaration of Helsinki, and governmental, state and local laws. Informed consent was obtained from each patient prior to study screening. The trial started in April 2013 and the last patient completed the last study visit September 2016.
Statistical analyses and sample size
Efficacy analyses were conducted in the modified intent-to-treat (mITT) population, defined as all subjects who received at least one dose of study drug. The primary end point was the proportion of responders to a composite SRI at week 52, the SRI-6, defined as: a ≥6 point reduction in SELENA-SLEDAI score compared with baseline; no new British Isles Lupus Assessment Group (BILAG) A organ domain scores or no more than 1 new BILAG B organ domain score at time of assessment compared with baseline and no worsening in Physician’s Global Assessment (PGA) score (<0.3 point increase from baseline on a 3-point scale). Subjects who required increases in background SLE medication, including increases in corticosteroid by >25% or 5 mg prednisone (or equivalent), new or increased SLE medications or withdrew from study for any reason were defined as treatment failures for all SRI variables. The primary end point compared SRI-6 response in the blisibimod arm to placebo using a two-sided test at the 5% level of significance at week 52. The primary end point was analysed using a logistic regression model with treatment group, region, race, baseline SELENA-SLEDAI score (≤12 vs >12), prednisone dose, immunosuppressant use and proteinuria category fitted as explanatory variables.
Based on observations at week 24 in a phase II study with blisibimod10 and abstract presentations of emerging week 52 data with the BAFF-targeted monoclonal antibody, tabalumab,3 4 SRI-6 responder rates at week 52 with drug and placebo were modelled to be 39% and 25%, respectively. Assuming these responder rates and a two-sided test for significance at an alpha level of 0.05, the power to detect a treatment difference for SRI-6 at week 52 for 400 subjects is 84%.
Secondary end points were analysed using the following methods: time-to-event variables were analysed using Cox proportional hazards regression models, proportion end points were analysed using logistic regression models, quantitative end points were analysed using analysis of covariance (ANCOVA) models and flare rate was analysed using a negative binomial regression model. All models included the same factors as used for the analysis of the primary efficacy end point. The ANCOVA models additionally included the baseline value of the corresponding parameter as a covariate. Secondary end points were to be tested in a prespecified hierarchical order, each at the 0.05 significance level. However, since the study null hypothesis was rejected due to failure of the primary efficacy analysis, all secondary analyses are considered to be exploratory and are reported here as such, without adjustment for multiplicity.
An interim futility analysis using the SRI-6 response at week 24 was conducted by an independent statistician after approximately 100 subjects completed 24 weeks of treatment or had withdrawn from study.
Results
Of the 588 subjects who were screened for this study, 442 were randomised, one of whom did not receive study drug (in the placebo arm) and was excluded from the mITT analyses (figure 1). The majority of subjects were female (93.7%) and of childbearing potential (average age 36.2 years), and the most common SLE organ manifestations based on SELENA-SELDAI score were dermal (95.7%), immunologic (90.7%) and musculoskeletal (79.8%). Approximately 29.9% of subjects had evidence of renal disease at enrolment, defined as UPCR ≥56.5 mg/mmol. The mean SELENA-SLEDAI score and corticosteroid dose were 13.5±4.2 and 15.6±9.1 mg, respectively. Demographics, baseline disease characteristics and SLE medications were approximately balanced between the two treatment arms (table 1). In general, the use of background SLE mediations in this study was similar in recent global clinical trials in SLE.1–4

Disposition of subjects in the CHABLIS-SC1 trial, including randomisation (5:4) into the treatment arms, numbers of subjects who completed study and reasons for discontinuation (and numbers of subjects).
Demographics and baseline disease characteristics
Effects on SRI, SELENA-SLEDAI and corticosteroid taper
The study did not meet the primary efficacy end point of SRI-6 at week 52 (46.9% with blisibimod, 42.3% in the standard-of-care control group, figure 2A). However, consistent trends from week 16 through week 52 were observed with blisibimod for both the SRI-6 and SRI-4 end points (figure 2B, C).

Effects of blisibimod (blue) and placebo (grey) on measures of systemic lupus erythematosus efficacy and corticosteroid taper in the modified intent-to-treat (mITT) population. SLE Responder Index -6 (SRI-6) data for week 52 (A) and at all visits (B), are plotted along with SRI-4 at all visits (C). The mean daily prednisone (or equivalent) dose is shown in (D), along with the proportion of subjects who achieved corticosteroid taper to prednisone ≤7.5 mg/day or equivalent at each visits after week 8 (the time after which steroid taper was encouraged) (E). In the evaluation of SRI-6 with steroid reduction (F), responders met all of the SRI-6 criteria week 52 while also having an oral corticosteroid dose from week 40 through week 52 that was lower than the dose on day 1. SRI analyses were conducted using a non-responder imputation method, wherein subjects were considered to be non-responders if they received new or increased corticosteroid or immunosuppressive or antimalarial medication relative to baseline, or used prohibited medication, or withdrew from study for any reason from the event date onwards. In all graphs, the proportion of subjects meeting the relevant responder criteria are reported relative to the number of subjects randomised to that treatment arm in the mITT or the relevant evaluable population. Statistical outcomes are annotated as *P<0.05 and **P<0.01.
The mean steroid dose after week 8 remained below baseline in the blisibimod group and above baseline in the placebo group, with the difference between groups slowly widening from ~3 mg/day prednisone at week 12 to ~5 mg/day at week 52 (figure 2D). Consistent with this observation, significantly more subjects in the blisibimod arm with a baseline prednisone dose of >7.5 mg/day or equivalent achieved corticosteroid taper to ≤7.5 mg/day at each visit from week 24 through week 52 (figure 2E), and more subjects on blisibimod achieved sustained corticosteroid taper over weeks 40 through 52 compared with placebo (17.2% vs 8.9%, P=0.019). A secondary analysis of the SRI-6 response was undertaken to examine whether the SRI-6 primary end point may have been confounded by imbalances in corticosteroid taper. In this end point, adapted from a recently published trial result,14 response was defined as meeting the week 52 SRI-6 criteria while also having an oral corticosteroid dose from week 40 through week 52 that was lower than the dose on day 1. With this analysis, 23.3% of blisibimod-treated subjects met the end point vs 14.3% of the control arm (P=0.056, figure 2F).
Effects on UPCR
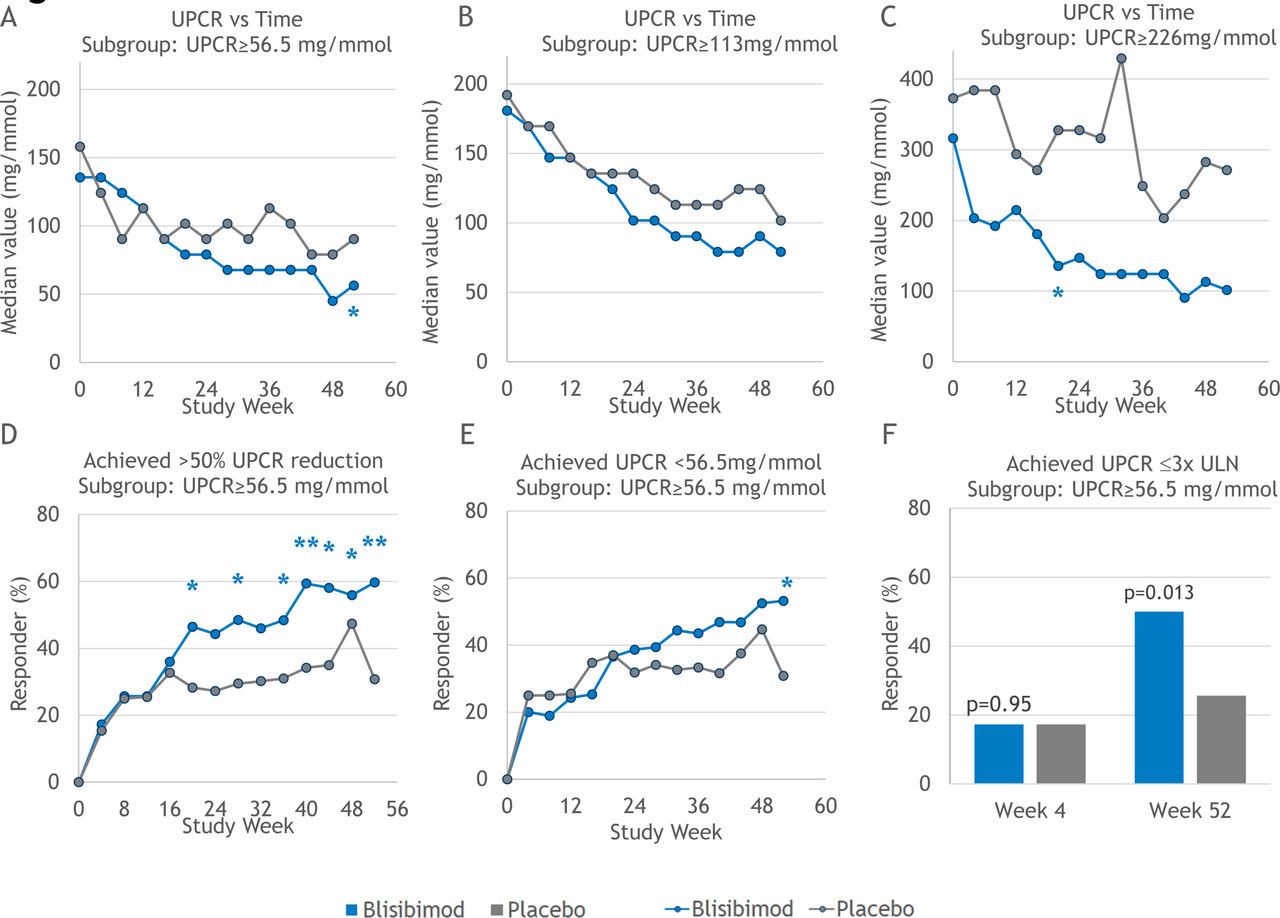
Treatment effects on UPCR were evaluated in subgroups of subjects defined by baseline UPCR ≥56.5 mg/mmol (n=135), ≥113 mg/mmol (n=82) and ≥226 mg/mmol (n=35). Within each subgroup, baseline UPCR levels were approximately balanced between treatment arms. Greater decreases in UPCR were observed among subjects randomised to blisibimod at all three ranges of proteinuria (figure 3A, B, C). The modest decreases from baseline in UPCR observed in subjects in the placebo arms of each of these subgroups may reflect the monthly clinical assessments and better adherence to therapy of subjects in trials compared with pretrial status. In the subgroup of subjects with baseline UPCR ≥56.5 mg/mmol, significantly more of the subjects who received blisibimod achieved >50% reduction in UPCR from baseline (figure 3D), and/or the threshold of UPCR <56.5 mg/mmol (figure 3E), and/or the threshold of UPCR ≤3 times the upper limit of normal (16.95 mg/mmol) at week 52 (figure 3F).

Effects of blisibimod (blue) and placebo (grey) on urinary protein:creatinine ratio (UPCR) in UPCR subgroups. Median UPCR were computed for both treatment arms in subgroups based on baseline UPCR: UPCR ≥56.5 mg/mmol (n=135, A), ≥113 mg/mmol (n=82, B) and ≥226 mg/mmol (n=35, C). Additional analyses in the subgroup of subjects with baseline UPCR ≥56.5 mg/mmol evaluated the proportion of subject who achieved >50% reduction in UPCR from baseline (D), and/or UPCR <56.5 mg/mmol (E), and/or UPCR ≤3 times the upper limit of normal (ULN, defined as 0.15 g/24 hours equivalent (F). For graphs A, B and C, data are shown as median value for all available observations from subjects in the relevant subgroup. Responder analyses in graphs D, E and F summarise the proportion of responder relative to number of subjects in the subgroup of subjects with baseline UPCR ≥56.5 mg/mmol. Statistical outcomes are annotated as *P<0.05 and **P<0.01.
Effects on blood and serum markers of B-cell activity or SLE disease activity
A trend towards decreased anti-dsDNA was observed with blisibimod (figure 4A), which was not statistically significant. Significant increases in complement C3 and C4 were observed with blisibimod compared with the standard-of-care control group (figure 4B, C) with the following changes in peripheral B-cell counts (detected by flow cytometry gated on side scatter and CD45, and expressed relative to concurrent lymphocyte counts): decreases in total B cells (CD20+CD19+), naïve B cells (CD20+CD19+IgD+CD27−), activated B cells (CD19+CD38+CD138+) (data not shown) and transient increases in the memory B cells (CD19+CD27+, figure 4D, E, F). Finally, significant decreases in mean concentrations of serum immunoglobulins IgM, IgG and IgA ranging from 10% to 26% (figure 4G, H, I), and anticardiolipin-directed IgG and IgM antibodies were observed with blisibimod compared with placebo. Five of the 245 (2.0%) subjects randomised to blisibimod and 3/197 (1.5%) subjects randomised to placebo were observed to have reduction in IgG to <4 g/L or lower. In all but two subjects, one in each treatment arm, the low IgG value was transient, recovering by the next observation. No infections were reported as serious adverse events in any subject with such reductions in IgG.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Treatment effects on SLE biomarkers, B cells and quantitative immunoglobulins. (A) Median change from baseline (BL) in serum concentrations of anti-dsDNA antibodies (mean at baseline 200.6±763.7 and 175.4±576.7 IU/mL for blisibimod and placebo), (B) mean change from BL in complement C3 (mean at baseline 0.881 and 0.885 g/L), (C) mean change from BL in C4 (mean baseline 0.174 and 0.175 g/L) are plotted for blisibimod (blue) and placebo (grey). Similarly, data are plotted for mean % changes from BL in B-cell counts determined by flow cytometry are reported as median per cent change from baseline for: (D) total B cells, (E) naïve B cells and (F) memory B cells (I), and mean % change from BL in serum concentrations of (D) immunoglobulin IgM (mean at baseline 1.15 and 1.07 g/L), (E) IgG (mean at baseline 14.53 and 14.94 g/L) and (F) IgA (mean at baseline 3.024 and 3.188 g/L). For all graphs, data are plotted using all available observations from subjects in the modified intent- to- treat population (*P<0.05, **P<0.01).
Effects on quality of life
At 3-month intervals throughout the study, subjects were asked to conduct a self-evaluation of their quality of life using the Lupus Quality of Life Questionnaire (Lupus QoL).15 To minimise bias, these evaluations were required to be conducted at study visit prior to any assessments of disease activity by the investigator or site staff. Modest improvements in the Lupus QoL total score were observed with blisibimod compared with control from week 12 onwards (P=ns).
Safety
Blisibimod was well tolerated, with similar numbers of subjects reporting treatment-emergent adverse events (TEAEs 69.8% vs 64.8%) and treatment-emergent serious adverse events (13.1% vs 17.3%) in the blisibimod and control arms, respectively. The only imbalances in reported TEAEs related to reactions at the injection site (erythema, reaction, rash, pain), which were reported more frequently with blisibimod than placebo (table 2); none was severe or serious. The latter imbalance also accounts in large part for the higher proportion TEAEs considered due to study drug (35.9% vs 23.5%), and TEAEs leading to discontinuation (5.3% vs 1.5%) on blisibimod.
Treatment-emergent adverse events
Discussion
Post hoc analyses of the combined datasets from two Phase III trials with belimumab, BLISS-52 and BLISS-76 demonstrated that better treatment benefit was detected among subjects with higher disease activity, seropositivity defined by anti-dsDNA high or complement C3/C4 low and ongoing corticosteroid use.5 These observations, corroborated by subsequent observations from the Phase III trials with tabalumab,4 provided the rationale for the design for the phase III CHABLIS-SC1 study, which prospectively evaluated a population of subjects with SLE enriched for the traits that, in earlier studies, were associated with greater treatment effect relative to standard-of-care alone.
The SRI-6 response rate in the control subjects in this study (42.3%) was very high compared with prior SLE trials conducted in subjects with baseline SELENA-SLEDAI scores of ≥6 (20.4%,1 29.2%,3 27.0%4) or ≥8 (33.7%16). Analysis of SRI-6 was based on a logistic regression model with the following covariates fitted as explanatory variables: region, SELENA-SLEDAI at baseline (<=12, >12), UPCR at screening (<226, ≥226 mg/mmol), prednisone dose at baseline (<median dose, ≥median dose) and immunosuppressant use at baseline (yes, no). None of these was found to significantly influence SRI-6 outcome (range of covariate P values: 0.26–0.81).
Secondary analyses of SRI-6 response provide possible explanations for the high response rates of control subjects. Specifically, when steroid reduction is included in the end point, analogous to the phase II SLE trial with anifrolumab,14 lower responder rates were observed and greater treatment effect with blisibimod. The secondary analyses of the effects of blisibimod on corticosteroid taper suggest that background corticosteroid use may have confounded the primary efficacy end point, since many of the control subjects received corticosteroid doses that were higher than baseline throughout the trial, averaging 3–5 mg/day higher than the blisibimod group, in which the mean dose was below baseline from week 12 onwards. Perhaps alternative methods of normalising corticosteroid use and taper between treatment arms might be considered in future SLE trials. Incorporation of steroid reduction within outcomes has also been found informative.14
The contribution of each organ system to improvements in the SELENA-SLEDAI was examined. The primary contributions to disease activity at baseline were, not surprisingly, from the musculoskeletal and mucocutaneous domains, but immunologic and renal manifestations were also substantial. Rapid improvements in both mucocutaneous and musculoskeletal disease activity were observed in both treatments arms at week 4 (>10%), week 8 (>25%), week 12 (>40%) and beyond. Responses in these domains can be sensitive to small changes in corticosteroids, and the higher steroid dosing in the placebo arm may have contributed to the relatively high response rates in that group.
In an analysis of subjects with baseline UPCR ≥56.5, ≥113 and/or ≥226 mg/mmol, reductions in proteinuria (UPCR) were observed with blisibimod. These observations, generated in subgroups of fewer than one-third of the subjects enrolled, support the hypothesis that blisibimod has therapeutic potential in subjects with renal lupus. Interpretation of these data is limited by the lack of renal biopsy data in most subjects, and by the exclusion of subjects at entry to the trial whose renal disease was deemed to require immediate initiation of new or increased immunosuppressive therapy. However, these findings are consistent with the observed treatment effects of blisibimod on anti-dsDNA autoantibodies, complement C3 and C4, serum immunoglobulins and B-cell lineages, which were similar to those reported previously for blisibimod9 10 and other BAFF inhibitors.1 2 These observations additionally confirm that subjects in the blisibimod arm achieved pharmacological inhibition of BAFF.
In summary, the CHABLIS-SC1 (see online supplementary file 1) trial prospectively evaluated an SLE population enriched for disease characteristics that had been previously associated with greater treatment effect, but failed to meet its primary end point of SRI-6. However, key secondary analyses of corticosteroid use and UPCR suggest that blisibimod might have important clinical benefits.
Supplementary file 1
Acknowledgments
The authors acknowledge the efforts and commitment of the patients, clinicians and study personnel who participated in the CHABLIS-SC1 trial. Portions of the data presented in this manuscript have been shared in summarised form as abstracts and presentations at the following conferences: European Congress of Rheumatology (EULAR), Madrid, Spain, June 2017, American College of Rheumatology (ACR), New Orleans, Louisiana, USA, November 2017 and American Society of Nephrology (ASN), San Diego California, USA, November 2017.
References
Footnotes
Handling editor Josef S Smolen
Contributors All authors provided substantial input to the design, conduct and/or analysis of this trial. In addition, all authors participated in the drafting of this manuscript and the responses to reviewers and approve this version for publication.
Funding This sponsorship of this clinical trial by Anthera Pharmaceuticals included all funding and oversight of all clinical operations, laboratory analyses, data collection, statistical analyses, drug manufacturing and distribution to conduct and complete the trials as described.
Competing interests JTM, MS, KCK, DW are current or prior consultants for the sponsor, Anthera Pharmaceuticals. MS was a clinical investigator in this trial. RSM and WRS are employees and shareholders of Anthera Pharmaceuticals.
Patient consent Detail has been removed from this case description/these case descriptions to ensure anonymity. The editors and reviewers have seen the detailed information available and are satisfied that the information backs up the case the authors are making.
Ethics approval Multiple EC/IRB bodies reviewed and approved this trial (in Belarus, Brazil, Colombia, Georgia, Guatemala, Hong Kong, Korea, Singapore, Malaysia, Mexico, Russia, Sri Lanka, Taiwan, Thailand and the Philippines).
Provenance and peer review Not commissioned; externally peer reviewed.