Article Text

Abstract
Objective The basic leucine zipper transcription factor, ATF-like (BATF), a member of the Activator protein-1 family, promotes transcriptional activation or repression, depending on the interacting partners (JUN-B or C-JUN). Here, we investigated whether the BATF/JUN complex exerts regulatory effects on catabolic and anabolic gene expression in chondrocytes and contributes to the pathogenesis of osteoarthritis (OA).
Methods Primary cultured mouse chondrocytes were treated with proinflammatory cytokines (interleukin-1β, IL-6 or tumour necrosis factor-α) or infected with adenoviruses carrying the Batf gene (Ad-Batf). Expression of BATF and JUN was examined in human and mouse experimental OA cartilage samples. Experimental OA in mice was induced by destabilisation of the medial meniscus or intra-articular injection of Ad-Batf. The chromatin immunoprecipitation assay was used to examine the binding of BATF and JUN to the promoter regions of candidate genes.
Results Overexpression of BATF, which forms a heterodimeric complex with JUN-B and C-JUN, induced upregulation of matrix-degrading enzymes and downregulation of cartilage matrix molecules in chondrocytes. BATF expression in mouse joint tissues promoted OA cartilage destruction, and conversely, knockout of Batf in mice suppressed experimental OA. Pharmacological inhibition of BATF/JUN transcriptional activity reduced the expression of matrix-degrading enzymes and protected against experimental OA in mice.
Conclusions BATF/JUN-B and BATF/C-JUN complexes play important roles in OA cartilage destruction through regulating anabolic and catabolic gene expression in chondrocytes. Our findings collectively support the utility of BATF as a therapeutic target for OA.
- Chondrocytes
- Osteoarthritis
- Cytokines
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Osteoarthritis (OA) is the most common form of degenerative arthritis associated with multiple pathological changes in whole joint tissues.1 During OA pathogenesis, mechanical stress and proinflammatory cytokines induce matrix-degrading enzymes and repress SOX9 in chondrocytes to induce cartilage destruction.2–4 Multiple signalling pathways stimulated by mechanical stress and proinflammatory signalling are integrated to activate or suppress the expression of the appropriate anabolic and catabolic genes.
Various extracellular stimuli promote the formation of unique dimeric transcription factor complexes that play pivotal roles in gene regulatory flexibility and enhance their DNA-binding specificity to facilitate tight control of gene expression.5 Activator protein-1 (AP-1) forms homodimers and heterodimers composed of JUN and FOS family members, which act as either oncogenic or tumour suppressor factors, depending on the specific dimer compositions.6 Many matrix metalloproteinase (MMP) promoters contain potential AP-1-binding sites, and AP-1 activation by cytokines, in turn, regulates MMP expression.7 For instance, interleukin (IL)-1β activates AP-1-mediated transcription in articular chondrocytes, resulting in induction of MMP13.8 Conversely, AP-1 stimulation by IL-1β leads to suppression of SOX9 and type II collagen in chondrocytes,9 implying that anabolic responses of chondrocytes are downregulated by IL-1β through AP-1 transcriptional regulation.
In this study, we examined the hypothesis that the basic leucine zipper transcription factor, ATF-like (BATF), a member of the AP-1 family, integrates signalling from mechanical stress and proinflammatory cytokines to regulate OA pathogenesis. BATF, which lacks a transactivation domain, forms a heterodimer with JUN proteins that binds to the AP-1 site and promotes transcriptional activation or repression.10–12 While BATF/JUN transcriptional complexes clearly regulate various biological functions, including development of immune cells,12 the specific role of BATF in OA pathogenesis has not been established to date.
Materials and methods
Human OA cartilage was sourced from individuals undergoing arthroplasty.13–15 Male wild-type (WT), Batf−/− (The Jackson Laboratory) and chondrocyte-specific Batf transgenic (TG) mice (Col2a1-Batf) generated using the Col2a1 promoter and enhancer13–15 were employed for experimental OA. Detailed experimental procedures are described in online supplementary materials and methods and supplementary table S1, including human and experimental OA, histology, immunohistochemistry, skeletal staining, primary culture of articular chondrocytes, adenoviruses, siRNA, reverse transcriptase (RT)-PCR, immunoblotting, immunoprecipitation, SOX9 reporter gene assay, chromatin immunoprecipitation (ChIP), AP-1 transcription factor assay and statistical analysis.
Supplemental material
Results
BATF and JUN are upregulated in cytokine-stimulated chondrocytes and OA cartilage
Treatment of chondrocytes with proinflammatory cytokines associated with OA pathogenesis (IL-1β, IL-6 and tumour necrosis factor (TNF)-α)4 led to increased BATF mRNA and protein levels. Among the BATF-binding partners, JUN-B was upregulated by all the cytokines and C-JUN enhanced by IL-1β only while JUN-D expression was not affected (figure 1A, B). BATF co-immunoprecipitated with JUN-B or C-JUN in chondrocytes stimulated with the cytokines (figure 1C, E). Expression of BATF was markedly elevated in OA-affected, damaged regions of human cartilage, compared with undamaged areas in the same patient (figure 1F). Moreover, levels of BATF and its binding partners, JUN-B and C-JUN, were upregulated in cartilage of mouse OA caused by destabilisation of the medial meniscus (DMM) (figure 1F), supporting a potential role of BATF/JUN-B and BATF/C-JUN complexes in OA pathogenesis.

Expression of basic leucine zipper transcription factor, ATF-like (BATF)/JUN-B or BATF/C-JUN complexes in chondrocytes and osteoarthritis (OA) cartilage. (A) qRT-PCR analyses of chondrocytes treated with interleukin (IL)-1β (6 h, 1 ng/mL), IL-6 (24 h, 100 ng/mL) or tumour necrosis factor (TNF)-α (6 h, 10 ng/mL). Values are means±SEM (*p<0.05, **p<0.001; n≥10). (B) Immunoblotting of BATF and its binding partners in chondrocytes stimulated with IL-1β, TNF-α or IL-6 (n≥4). Coimmunoprecipitation of BATF with JUN-B and C-JUN after stimulation with IL-1β (C), IL-6 (D) or TNF-α (E) (n≥4). (F) Alcian blue and safranin-O staining and immunostaining for BATF, JUN-B and C-JUN in human and destabilization of the medial meniscus-operated mouse OA cartilage (n≥6). Scale bar: 50 μm.
BATF regulates catabolic and anabolic gene expression in chondrocytes
Overexpression of BATF in chondrocytes via Ad-Batf infection led to increased mRNA and protein levels of the matrix-degrading enzymes, MMP3, MMP13 and ADAMTS5 (figure 2A, B), which play crucial roles in OA cartilage destruction.16–18 BATF overexpression additionally elevated JUN-B and C-JUN, but not JUN-D protein levels (figure 2B). Conversely, expression levels of cartilage matrix type II collagen and aggrecan and expression/transcriptional activity of SOX9 were markedly decreased (figure 2A–C).19 ,20 Knockdown of Batf using specific siRNA blocked upregulation of MMP3 and MMP13 by IL-6, but not IL-1β and TNF-α (figure 2D, E). The differential effects of these cytokines may be attributed to the distinct signalling pathways activated in chondrocytes. IL-1β and TNF-α activate AP-1 and also nuclear factor (NF)-κB, which promote MMP expression.21 Therefore, inhibition of AP-1 via Batf knockdown may be insufficient to block IL-1β-induced or TNF-α-induced expression of MMPs owing to the simultaneous activation of NF-κB.
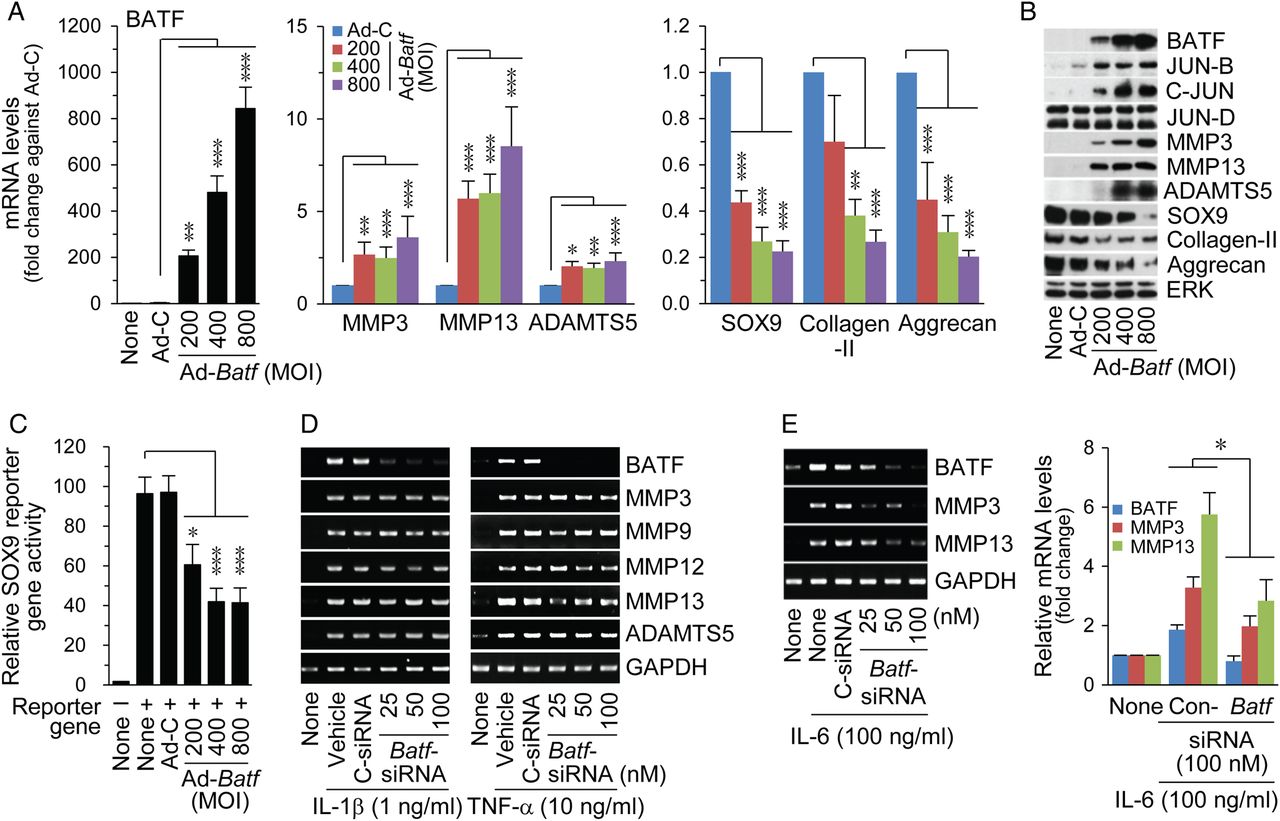
BATF (basic leucine zipper transcription factor, ATF-like) regulates catabolic and anabolic genes in chondrocytes. qRT-PCR (A, n=10) and western blot analysis (B, n=5) of catabolic and anabolic factors in chondrocytes infected with an indicated multiplicity of infection (MOI) of control adenovirus (Ad-C) or Ad-Batf. (C) SOX9 reporter gene assay in chondrocytes infected with adenoviruses (n=6). Chondrocytes were transfected with control siRNA (C-siRNA) or Batf siRNA, and exposed to interleukin (IL)-1β, tumour necrosis factor (TNF)-α (D) or IL-6 (E). mRNAs were detected using RT-PCR and quantified with qRT-PCR (n=8). Values represent means±SEM (*p<0.05, **p<0.01, ***p<0.0005).
ChIP assays on BATF-overexpressing chondrocytes revealed that the BATF/JUN-B complex interacts with a BATF-binding motif (5′-TGAGT[G/A]-3′) in the promoter region of Mmp13, whereas the BATF/C-JUN complex binds to promoters of Mmp3, Adamts5 and Sox9 (figure 3A), suggesting direct modulation of these genes by BATF/JUN complexes. Moreover, BATF bound to the promoter regions of Junb and Jun, indicating direct upregulation of its JUN partners in chondrocytes (figure 3B). BATF additionally interacted with the promoters of Mmp3, Mmp13, Adamts5 and Sox9 in chondrocytes stimulated with IL-1β, whereas TNF-α stimulated BATF binding to Mmp3, Mmp13 and Sox9 and IL-6 to Mmp3 and Mmp13 (see online supplementary figure S1).

Regulatory mechanisms of the BATF (basic leucine zipper transcription factor, ATF-like)/JUN complex affecting catabolic and anabolic gene expression in chondrocytes. (A and B) Chromatin immunoprecipitation (ChIP) assay for binding of BATF, JUN-B or C-JUN in chondrocytes infected with Ad-Batf (800 MOI). Normal rabbit IgG was used as the negative control (n≥4). (C) Whole-cell lysates from chondrocytes infected with Ad-Batf (800 MOI) were co-immunoprecipitated with anti-C-FOS antibody or control IgG, and immunoblotted with anti-C-JUN antibody. (D) Western blot of IRF4 in chondrocytes stimulated with interleukin (IL)-1β, IL-6 and tumour necrosis factor (TNF)-α or infected with Ad-Batf. (E) ChIP assay for IRF4-binding in chondrocytes infected with Ad-Batf (n≥4).
BATF was initially identified as a dominant-negative regulator of C-FOS/C-JUN-mediated transcription.11 However, its overexpression did not influence C-FOS/C-JUN complex formation in chondrocytes in our experiments (figure 3C). BATF/JUN also forms ternary complexes with interferon regulatory factor (IRF)-4 and IRF8 that bind to different types of AP-1/IRF composite elements.22–24 Stimulation of chondrocytes with IL-1β, IL-6 or TNF-α and BATF overexpression did not affect IRF4 protein levels (figure 3D), whereas IRF8 was not detectable under our experimental conditions (data not shown). However, ChIP assays revealed that IRF4 interacts with the same AP-1-binding sites of Mmp3 and Adamts5 promoters (figure 3E) at which BATF and C-JUN bind together (figure 3A), suggesting that the BATF/C-JUN/IRF4 complex regulates expression of Mmp3 and Adamts5. In contrast, IRF4 did not interact with promoters of Mmp13 and Sox9 (figure 3E). Indeed, the BATF-binding sites on the promoter regions of Mmp3 and Adamts5 contain an IRF4-binding motif (GAAA) juxtaposed with the BATF-binding motif, which is not present in the BATF-binding sites of Mmp13 and Sox9, suggesting that variations in IRF4 binding are related to promoter-specific differences.
AP-1 transcriptional activity is also regulated by mitogen-activated protein (MAP) kinases.25 Among the MAP kinase subtypes, C-JUN N-terminal kinase (JNK) is particularly important for direct AP-1 activation via phosphorylation of C-JUN.26 Indeed, pharmacological inhibition of JNK, but not p38 MAP kinase, repressed BATF-mediated transcriptional activation (see online supplementary figure S2). These results indicate that JNK regulates BATF/C-JUN-mediated expression of MMP3 and ADAMTS5 in chondrocytes.
Overexpression of BATF in joint tissues promotes OA pathogenesis
The role of BATF in OA pathogenesis was examined via intra-articular (IA) injection of Ad-Batf into mouse knee joints. Efficient adenoviral infection in mouse joint tissue was confirmed by expression of green fluorescence protein (GFP) via IA injection of adenovirus carrying the GFP gene (Ad-Gfp) (see online supplementary figure S3A). Ad-Batf injection effectively induced BATF overexpression in all joint tissues (see online supplementary figure S3B), in turn, causing cartilage destruction (figure 4A) and synovial inflammation (figure 4B), with concomitant upregulation of MMP3 and MMP13 and downregulation of SOX9 in cartilage tissue (figure 4C).

Overexpression of basic leucine zipper transcription factor, ATF-like (BATF) in cartilage induces osteoarthritis in mice. Cartilage destruction (A), synovial inflammation (B) and immunostaining of BATF, MMP3, MMP13 and SOX9 (C) in joint sections of wild-type (WT) mice intra-articular-injected with Ad-C or Ad-Batf (n=10). (D) Safranin-O staining and Osteoarthritis Research Society International (OARSI) grade in 12-month-old WT and Col2a1-Batf transgenic (TG) mice (n=10). (E) Cartilage destruction in sham-operated and destabilization of the medial meniscus (DMM)-operated WT and Col2a1-Batf TG mice (n=9). Values represent means±SEM (*p<0.05, **p<0.005, ***p<0.0005). Scale bar: 50 μm.
To examine the cartilage-specific function of BATF, we generated Batf TG mice using Col2a1 promoter and enhancer (see online supplementary figure S4A). Compared with age-matched WT mice, Batf TG mice exhibited spontaneous cartilage destruction at 12 months (figure 4D). However, only a small degree of cartilage destruction was evident, suggesting that chondrocyte-specific BATF minimally affects ageing-associated OA. In contrast, cartilage destruction via DMM surgery was significantly enhanced in Batf TG mice (figure 4E), indicating that BATF in cartilage tissue enhances post-traumatic OA-related destruction.
Genetic ablation of Batf suppresses OA pathogenesis
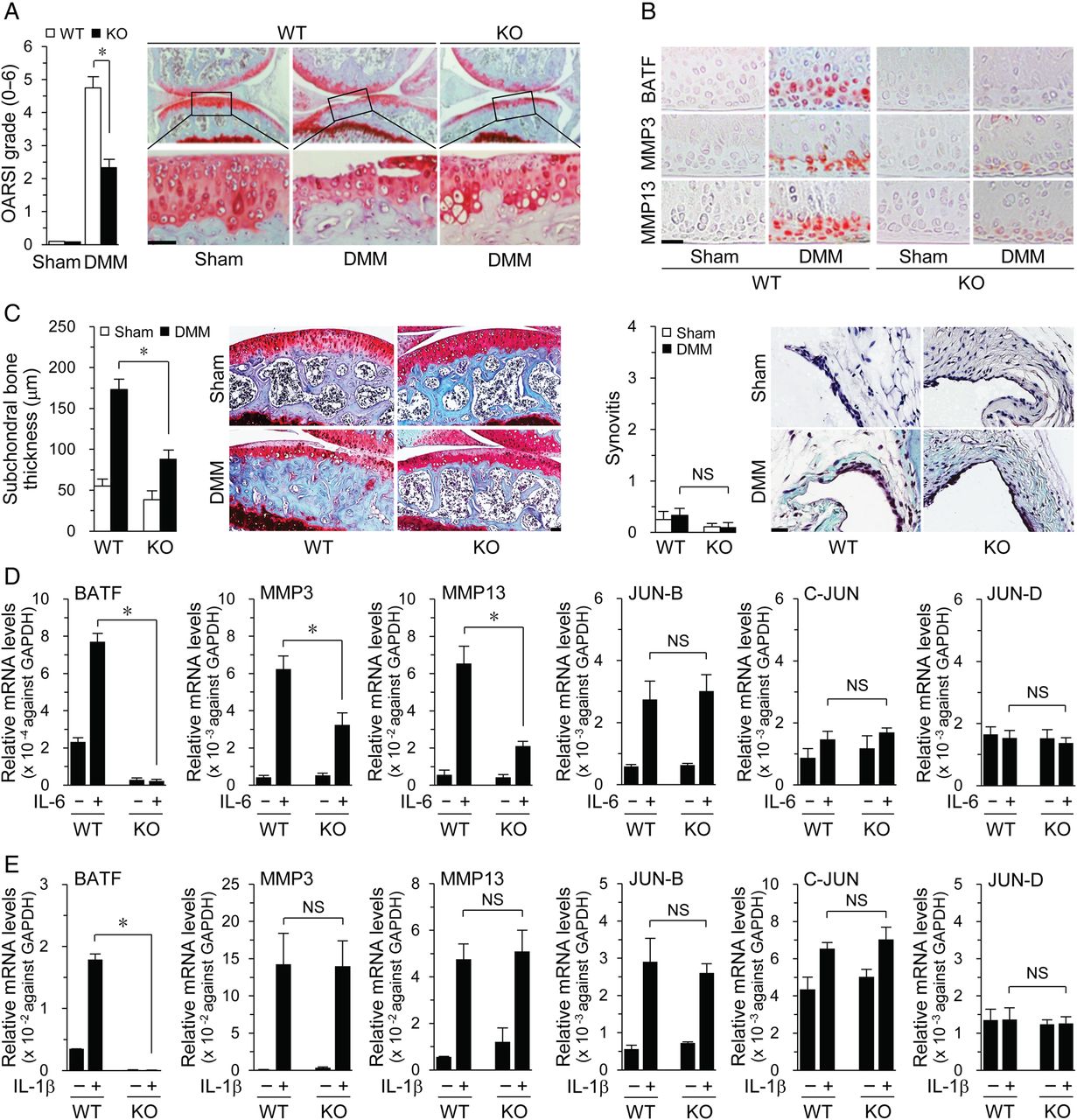
Next, we used Batf−/− mice, which were viable and developed normally27 with no defects in bone, cartilage or limb development (see online supplementary figure S5). Notably, cartilage destruction was significantly reduced in DMM-operated Batf−/− mice, compared with that in WT mice, along with reduced MMP3 and MMP13 expression (figure 5A, B). Subchondral bone sclerosis was also decreased in DMM-operated Batf−/− mice whereas synovial inflammation was not induced by DMM in both WT and Batf−/− mice (figure 5C). These results indicate a requirement for BATF in post-traumatic OA pathogenesis. In addition, IL-6-induced expression of MMP3 and MMP13 was suppressed in Batf−/− chondrocytes (figure 5D), whereas IL-1β-induced MMP3 and MMP13 levels remained unchanged (figure 5E), suggesting that IL-1β promotes MMP expression via BATF-independent pathways. Interestingly, while BATF overexpression induced JUN-B and C-JUN (figure 2B), IL-1β-induced or IL-6-induced JUN-B, C-JUN and JUN-D levels remained unchanged in Batf−/− chondrocytes (figure 5D, E), suggestive of BATF-independent stimulation of JUN protein expression by these cytokines.
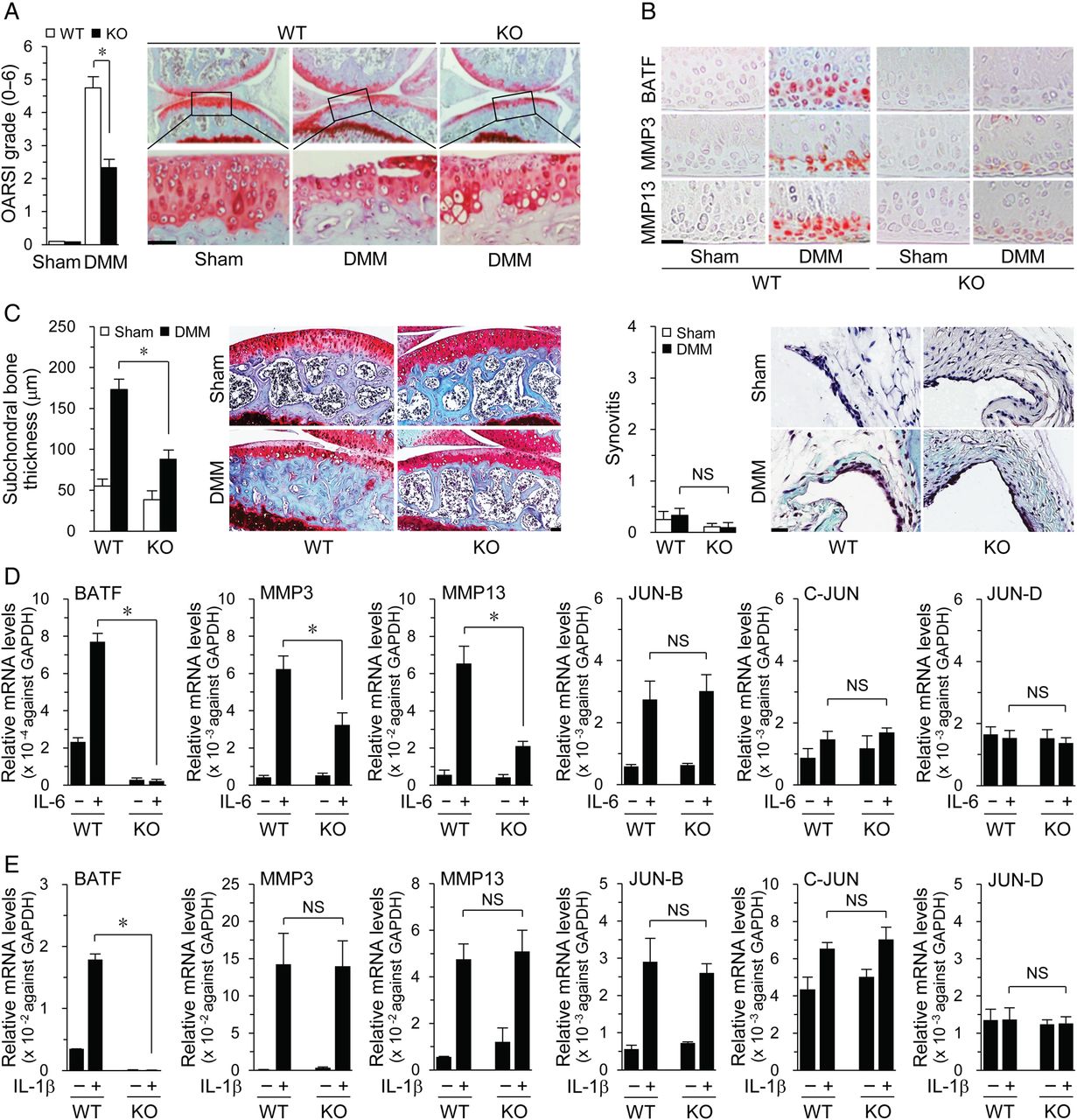
Batf (basic leucine zipper transcription factor, ATF-like) knockout (KO) in mice suppresses destabilization of the medial meniscus (DMM)-induced osteoarthritis. Cartilage destruction (A) and immunostaining of the indicated targets (B) in cartilage of sham-operated and DMM-operated wild-type (WT) and Batf−/− mice (n=7 mice). (C) Subchondral bone sclerosis and synovial inflammation in sham-operated and DMM-operated WT and Batf−/− mice (n=7 mice). Chondrocytes isolated from Batf−/− and WT littermates were treated with interleukin (IL)-6 (100 ng/mL) (D) or IL-1β (1 ng/mL) (E). qRT-PCR analyses of mRNA levels of the indicated genes (n=8). Values represent means±SEM (*p<0.001). NS, not significant. Scale bar: 50 μm.
Inhibition of the BATF/JUN complex suppresses experimental OA in mice
To explore the potential of BATF as a therapeutic target of OA, we examined whether inhibition of BATF/JUN affects catabolic factor expression and subsequent OA pathogenesis. To this end, we employed the benzophenone derivative, T-5224, which prevents DNA binding of C-FOS/C-JUN to the AP-1-binding motif, TPA-responsive element (TRE) (5′-TGAGTCA-3′), and inhibits C-FOS/C-JUN-mediated transcription.28 T-5224 effectively inhibited the binding of BATF, JUN-B and C-JUN proteins to TRE (figure 6A) and significantly abrogated upregulation of MMP3, MMP13 and ADAMTS5 induced by BATF in chondrocytes (figure 6B). Consistently, IA injection of T-5224 into mouse knee joints inhibited OA pathogenesis, including cartilage destruction and subchondral bone sclerosis triggered by DMM surgery (figure 6C) or IA injection of Ad-Batf (figure 6D). Our results support the utility of the BATF/JUN complex as an effective therapeutic target for OA.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Inhibition of BATF (basic leucine zipper transcription factor, ATF-like (BATF))/JUN complexes suppresses osteoarthritis pathogenesis. Activator protein-1 (AP-1) binding activity (A) and mRNA levels (B) in chondrocytes infected with Ad-C or Ad-Batf (800 MOI) without or with T-5224 (20 μM) (n=6). Cartilage destruction and subchondral bone sclerosis in sham-operated and destabilization of the medial meniscus (DMM)-operated wild-type (WT) mice (C) or WT mice intra-articular injected with Ad-C or Ad-Batf (D) in combination with T-5224 or vehicle (n=8). Values represent means±SEM (*p<0.05, **p<0.01, ***p<0.001). Scale bar: 50 μm.
Discussion
We initially identified BATF as an upregulated transcription factor in chondrocytes stimulated with IL-1β, IL-6 and TNF-α, the major proinflammatory cytokines in OA pathogenesis. IL-1β is associated with cartilage destruction and TNF-α with driving the inflammatory cascade.4 IL-6 has been shown to induce MMP3 and MMP13 protein expression, resulting in OA cartilage destruction in mice.15 In view of the finding that these cytokines enhance BATF expression in chondrocytes, we investigated the specific functions of BATF in OA pathogenesis. Our gain-of-function (IA injection of Ad-Batf and Batf TG mice) and loss-of-function (Batf KO mice and IA injection of AP-1 inhibitor) studies clearly indicate that BATF is a catabolic regulator of OA pathogenesis. We employed conventional Batf−/− rather than cartilage-specific KO mice for the experiments since OA is considered a whole-joint disease involving multiple pathological changes in different cells of joint tissue.1 Therefore, deletion of Batf in whole-joint tissues may be a more suitable approach for investigating BATF functions in OA joints.
Our results collectively demonstrate that BATF promotes OA by upregulating catabolic genes and downregulating anabolic genes in chondrocytes. This capacity of BATF appears to be dependent on specific interacting partners and activation of the signalling cascade. For instance, the BATF/JUN-B complex regulates Mmp13, whereas the BATF/C-JUN complex regulates Mmp3, Adamts5 and Sox9. This finding is consistent with previous studies showing that JUN-B regulates MMP13 expression in endothelial cells29 and C-JUN regulates MMP3 expression in synoviocytes.30 C-JUN overexpression is also associated with inhibition of SOX9 transcriptional activity and reduction of type II collagen expression in chondrocytes.9 Thus, BATF/C-JUN appears to suppress SOX9 expression, thereby reducing SOX9-dependent transcription in chondrocytes. BATF also interacts with IRF4 and C-JUN to form a ternary complex that regulates expression of Mmp3 and Adamts5. Our results indicate that JNK activation is necessary for BATF/C-JUN-mediated transcription in chondrocytes. While BATF overexpression led to upregulation of JUN-B and C-JUN, Batf KO in chondrocytes did not affect IL-1β-induced or IL-6-induced JUN-B or C-JUN expression, indicating that Batf is sufficient but not necessary for Junb and Jun expression in chondrocytes. Presumably, transcription factors or signalling pathways other than BATF may also mediate IL-1β-induced and IL-6-induced expression of JUN-B and C-JUN.
Another interesting finding is that ageing-associated OA cartilage destruction in Batf TG mice is mild, compared with that triggered by DMM surgery or IA injection of Ad-Batf. The minimal effects of BATF overexpression in aged Batf TG mice may be attributable to expression of BATF-binding partners (JUN-B and C-JUN). Overexpression of BATF via Ad-Batf increases JUN-B and C-JUN protein levels in chondrocytes. Interestingly, however, JUN-B and C-JUN levels are not altered in chondrocytes of Batf TG mice. This may be due to the relatively low expression of BATF in chondrocytes of Batf TG mice (8.7-fold increase; online supplementary figure S4D) compared with that in Ad-Batf-infected chondrocytes (∼800-fold increase; figure 2A). Alternatively, it is possible that BATF induction by proinflammatory cytokines is accompanied by mechanosensitive induction of JUN-B and C-JUN to initiate cartilage destruction in Batf TG mice. Indeed, dynamic compression of cartilage explants and chondrocytes embedded in agarose gel is known to induce C-JUN and JUN-B expression in chondrocytes.3 ,31 In Batf TG mice, joint instability caused by DMM additively induces JUN expression in chondrocytes, thus enhancing the transcriptional activity of the BATF/JUN complex and subsequent cartilage destruction. Therefore, it is likely that proinflammatory signalling and mechanical stress synergistically promote cartilage destruction via BATF/JUN complexes. We additionally observed that cartilage-specific Batf TG mice exhibit normal skeletal development. In addition to cartilage, BATF is overexpressed in proliferating chondrocytes of growth plates in Batf TG mice. However, similar to data obtained with cartilage in Batf TG mice, expression levels of BATF-binding partners (JUN-B and C-JUN) and downstream target genes (MMP3 and MMP13) were not significantly different between growth plates of WT and Batf TG mice (see online supplementary figure S4). Again, this may be due to the relatively low expression of BATF in chondrocytes of Batf TG mice, compared with that in Ad-Batf-infected chondrocytes.
BATF is reported to directly regulate IL-17A and IL-17F required for differentiation of Th17 cells.27 CD4+ T cells infiltrate the sublining layer of the human OA synovium.32 Furthermore, IL-17 signalling induces MMP13 production in OA chondrocytes.33 However, BATF overexpression did not induce IL-17A or IL-17F in primary cultured chondrocytes, fibroblast-like synoviocytes, cartilage or synovial tissues in our study (see online supplementary figure S6A,B), suggesting that the protein is dispensable for IL-17-mediated inflammatory responses in OA joints.
Our results collectively support the utility of BATF as a therapeutic target for OA pathogenesis. The AP-1 inhibitor, T-5224, was an effective blocker of BATF/JUN-mediated transcriptional activation, leading to suppression of AP-1-mediated expression of MMP3 and MMP13. Furthermore, T-5224 inhibited OA pathogenesis triggered by Ad-Batf infection and DMM surgery, supporting its potential as a candidate to prevent OA pathogenesis.
References
Footnotes
Handling editor Tore K Kvien
Contributors JR: Study design, data acquisition, data analysis and interpretation, manuscript preparation and approval; S-HP, S-KP, J-HK and C-WH: data acquisition, data analysis and manuscript approval; C-HC provided and evaluated human joint samples. J-SC (jschun@gist.ac.kr) takes responsibility for the integrity of this work.
Funding This work was supported by grants from the Korea Health Technology R&D Project (HI14C3484 and HI16C0287) through the Korea Health Industry Development Institute (KHIDI) funded by the Ministry of Health & Welfare, the National Research Foundation of Korea (2012R1A1A2044648) and by the GIST Research Institute (GRI).
Competing interests None declared.
Patient consent Obtained.
Ethics approval The Institutional Review Board of the Wonkwang University Hospital approved the use of human materials. All animal experiments were approved by the Gwangju Institute of Science and Technology Animal Care and Use Committee.
Provenance and peer review Not commissioned; externally peer reviewed.