Article Text

Abstract
Objectives Interferon-γ (IFNγ) is the pivotal mediator in murine models of primary haemophagocytic lymphohistiocytosis (pHLH). Given the similarities between primary and secondary HLH (sec-HLH), including macrophage activation syndrome (MAS), we investigate the involvement of the IFNγ pathway in MAS by evaluating levels of IFNγ and of the induced chemokines, and their relation with laboratory parameters of MAS in systemic juvenile idiopathic arthritis (sJIA) patients with MAS and in a murine MAS model.
Methods The Luminex multiplexing assay was used to assess serum levels of interleukin (IL)-1β, IL-6, IFNγ and of the IFNγ-induced chemokines CXCL9, CXCL10 and CXCL11 in patients with sec-HLH (n=11) and in patients with sJIA (n=54), of whom 20 had active MAS at sampling. Expression of IFNγ-induced chemokines was assessed in IL-6 transgenic mice in which MAS is induced by TLR4 stimulation with lipopolysaccharide.
Results Levels of IFNγ and of IFNγ-induced chemokines were markedly elevated during active MAS and sec-HLH and were significantly higher in patients with MAS compared with active sJIA without MAS. Levels in patients with active sJIA without MAS were comparable to those of patients with clinically inactive sJIA. During MAS, ferritin and alanine transferase levels and neutrophil and platelet counts were significantly correlated with serum levels of IFNγ and CXCL9. In murine MAS, serum levels of ferritin were significantly correlated with mRNA levels of Cxcl9 in liver and spleen.
Conclusions The high levels of IFNγ and of IFNγ-induced chemokines and their correlation with the severity of laboratory abnormalities of MAS suggest a pivotal role of IFNγ in MAS.
- Inflammation
- Cytokines
- Chemokines
- Juvenile Idiopathic Arthritis
Statistics from Altmetric.com
Introduction
The term macrophage activation syndrome (MAS) defines a severe, potentially fatal complication of rheumatic diseases. It occurs typically in the context of systemic juvenile idiopathic arthritis (sJIA) with 10–20% of patients developing MAS during disease course. In sJIA, MAS occurs typically during active disease, frequently at onset. Features of MAS include fever, splenomegaly, haemorrhages and liver, central nervous system and kidney involvement, and eventually multiple organ failure. Laboratory abnormalities include decrease in white blood cells, platelet and haemoglobin, hypertransaminasemia, hyperferritinemia and evidence of intravascular coagulation.1 MAS causes significant morbidity and mortality accounting for a significant portion of the deaths due to sJIA.2 ,3
MAS shares clinical and laboratory features with haemophagocytic lymphohistiocytoses (HLH) and is indeed currently classified among secondary HLH (sec-HLH).4 Primary HLH (pHLH) is caused by mutations of genes coding for proteins involved in granule exocytosis, including PRF1, UNC13D, STXBP2, STX11, RAB27A and XIAP, typically leading to defective cytotoxic activity of CD8+ and Natural Killer (NK) cells. In the absence of an identifiable genetic cause and/or familial inheritance, HLH is defined as secondary. Sec-HLH can occur in the absence of a demonstrable trigger or in the context of infections, malignancies or rheumatic diseases, the latter being commonly referred to as MAS. The genetic basis of MAS is being progressively unravelled, with a number of studies pointing to the association of MAS, and in general of sec-HLH, with heterozygosity for low-penetrance mutations of the same pHLH causative genes.5–9 These findings suggest a shared mechanism.
Studies in patients with pHLH and murine models of pHLH support the hypothesis that defective cytotoxic activity and abnormalities in antigen-presenting cell-CD8+ T cell crosstalk lead to T cell hyperactivation and defective silencing of the immune response. These result in uncontrolled immune activation and hyperproduction of pro-inflammatory cytokines by T lymphocytes and macrophages, leading to organ damage. Studies in perforin-deficient and Rab27-deficient mice point to a critical role of interferon-γ (IFNγ) produced by activated CD8+ T cells. In perforin-deficient mice, neutralisation of IFNγ leads to survival in an otherwise lethal syndrome, with reversal of biochemical and haematological abnormalities.10 ,11 In Rab27-deficient mice, neutralisation of IFNγ leads to marked improvement of the involvement of target organs, including central nervous system.11 High circulating IFNγ levels are found in patients with HLH:12–15 these studies included a significant, although variable, proportion of patients without a demonstrable genetic cause.12–15
We report that levels of IFNγ and of chemokine (C-X-C motif) ligand 9 (CXCL9), CXL10 and CXCL11, three chemokines that are known to be induced by IFNγ, are elevated in patients with MAS complicating sJIA, but not in patients with active sJIA without MAS. Levels of IFNγ, CXCL9, CXCL10 and CXCL11 correlated with laboratory parameters of disease in patients with MAS and in a MAS murine model.
Materials and methods
Patients
Peripheral blood was collected from patients with sJIA with or without MAS in three paediatric rheumatology centres: Ospedale Bambino Gesù (Rome), Istituto Gaslini (Genoa) and Cincinnati Children's Hospital. Fifty-four patients with sJIA (age at onset 7.9 years, IQR 4.6–13.6 years; female 48%) who met the International League of associations for Rheumatology (ILAR) classification criteria for sJIA were studied.16 For 20 patients, at least one sample was collected during MAS episodes, as diagnosed by the treating physicians. Twenty-eight patients with active sJIA without evidence of MAS had samples available. 17 out of 20 (85%) MAS met the newly proposed classification criteria and one of the patients with active sJIA did (3.6%)17 (see online supplementary table S1). Thirty-five samples were available from 35 patients with sJIA (both with or without MAS in their history) during clinically inactive disease.18
supplementary table S1
Items of diagnostic guidelines or classification criteria present in the patients with sec-HLH or MAS in sJIA included in the study, recruited in Rome (sec-HLH=11, sJIA with or without MAS=22), Genoa (sJIA with or without MAS=10) and Cincinnati (sJIA with or without MAS=22). The Table shows also the presence of each individual criterion at time of sampling in patients divided according to those who did not yet receive any treatment (Off therapy) and those who had already received one treatment for MAS/HLH including glucocorticoid pulses, cyclosporine-A, anakinra or cyclophosphamide (On therapy).
Since IFNγ is reported to be increased in sec-HLH, samples were collected from 11 patients with sec-HLH (age at onset 8.6 years, IQR 4.1–12.9 years; female 36%) and used as positive controls. All patients with sec-HLH met the 2004-HLH diagnostic guidelines19 (see online supplementary table S1). A diagnosis of genetic HLH was excluded based on absence of family history, absence of mutations in the genes known to cause HLH and normal functional studies (NK activity, perforin expression, CD107 degranulation).
Of the 20 patients sampled during active MAS, 7 were not receiving any treatment; 13 had already received treatments for MAS or sec-HLH, including glucocorticoid pulses, ciclosporin A, anakinra or cyclophosphamide. Also, 6/11 patients with sec-HLH were not receiving specific treatment at sampling, while the remaining 5 patients had already received at least one of the above-mentioned treatments. The clinical and laboratory features of MAS were present in patients who had already received treatment (see online supplementary table S1).
Quantification of cytokines
Levels of interleukin (IL)-6, IL-1β, IFNγ, CXCL9, CXCL10 and CXCL11 were analysed by Luminex multiplexing technology. All reagents were provided with Milliplex MAP kits (Millipore) and were prepared according to the manufacturer’s protocol. The 25 μL/well of standards, blank and quality check samples were added in duplicate in Milliplex MAP 96-well plates, followed by addition of 25 μL of serum matrix. And, 25 μL of assay buffer was added to each well followed by the addition of 25 μL of samples in duplicate or triplicate, depending on available volume. The plate was measured on the Luminex 200 system (Luminex).
Animal experiments
The phenotype of the IL-6 transgenic mice and the features of the MAS-like syndrome induced by administration of toll-like receptor (TLR) ligands have been described.20 Mice were maintained under specific pathogen-free conditions and handled in accordance with national policies. The protocol was approved by the local authority. Mice (age 10–14 weeks) were administered intraperitoneally one dose of 5 μg/g body weight of lipopolysaccharide (LPS, Escherichia coli serotype 055:B5; Sigma-Aldrich) and sacrificed after 30 hours. Total RNA was extracted from spleen and liver using Trizol (Life Technologies). cDNA was obtained using the Superscript Vilo kit (Invitrogen). Real-time PCR was performed using the TaqMan Universal PCR Master Mix (Applied Biosystems) with the mouse Cxcl9 and Cxcl10 gene expression assays (Applied Biosystem). Expression data were normalised using Gapdh (Applied Biosystem). Data are expressed as arbitrary units, determined using the 2−Δct method. Serum ferritin concentrations were determined using a commercially available ELISA (ALPCO Diagnostics).
Statistical analysis
Statistical analysis was performed using GraphPad Prism 5. Continuous variables were expressed as medians and IQRs and were compared using the Mann–Whitney U test. The Wilcoxon signed-rank test was used to compare two paired groups. We used Spearman rank correlation to assess the relation with laboratory parameters. A p value <0.05 was considered significant.
Results
Increased levels of IFNγ and IFNγ-induced chemokines in MAS
As expected,21 IL-6 levels were significantly higher (p<0.01) in patients with active sJIA compared with patients with clinically inactive disease (table 1). As reported in previous studies, serum IL-1β levels were below the limit of detection in the majority of patients with active sJIA. Notably, there were no differences in the levels of IFNγ and of the three IFNγ-induced chemokines among patients with clinically active sJIA and patients with clinically inactive disease.
Serum levels of interleukin (IL)-1β, IL-6, interferon γ (IFNγ) and of the three IFNγ-related chemokines CXCL9, CXCL10 and CXCL11 in patients with active secondary haemophagocytic lymphohistiocytosis (sec-HLH), with active macrophage activation syndrome (MAS) at sampling, with active systemic juvenile idiopathic arthritis (sJIA) without MAS at sampling and with clinically inactive sJIA
Levels of IL-1β and IL-6 were comparable in patients with MAS at sampling and in patients with active sJIA without MAS, showing that the levels of the two cytokines known to play a pivotal role in sJIA do not increase during full-blown MAS (table 1). In contrast, circulating IFNγ levels were significantly higher in patients with active MAS compared with patients with active sJIA without MAS at sampling. Similarly, levels of CXCL9, CXCL10 and CXCL11 were also markedly higher in patients with active MAS (table 1); notably CXCL9 median levels were approximately 15-fold higher in patients with MAS compared with patients without MAS. In patients with sec-HLH, levels of IFNγ, as well as levels of the three IFNγ-induced chemokines, were markedly increased (table 1) and not significantly higher than those of patients with MAS. In patients with MAS and in patients with active sec-HLH, the levels of IFNγ and of the three IFNγ-induced chemokines were comparable independently of the presence or absence of treatment (not shown).
Levels of IFNγ and of the IFNγ-induced chemokines in individual patients from whom paired samples were available were significantly higher in samples obtained during MAS (figure 1). In several patients, samples were available prior to and after MAS, and demonstrate that IFNγ and IFNγ-induced chemokines levels return to normal upon resolution of MAS. For example, one patient had three MAS episodes, with samples obtained during episodes and during phases without MAS: elevated levels of IFNγ and of the three IFNγ-induced chemokines were found only during MAS (figure 2).

Serum levels of interferon γ (IFNγ) and of CXCL9, CXCL10 and CXCL11 in individual patients from whom paired samples were available during active macrophage activation syndrome (MAS) and during active systemic juvenile idiopathic arthritis (sJIA) without MAS at sampling (Act sJIA). Significance levels (p) were obtained using the Wilcoxon rank test for paired samples.

Changes in white blood cell (WBC) and platelet counts and in ferritin levels (A) and changes in serum levels of interferon γ (IFNγ), CXCL9, CXCL10 and CXCL11 (B) in one patient who presented, during the course of his systemic juvenile idiopathic arthritis, three episodes of macrophage activation syndrome (MAS).
Notably, during active sJIA without MAS at sampling, levels of CXCL9 (median 1530, IQR 870–6065) and CXCL10 (649, 290–1488) were significantly higher in patients with a history of MAS compared with those of patients without a history of MAS (651, 420–1129; 220, 159–512, respectively) (see online supplementary table S2). This finding leads to the hypothesis that patients with a history of MAS may produce more CXCL9, and this may be the consequence of a subclinical activation of IFNγ production.
supplementary table S2
Circulating levels of IL-1β, IL-6, IFNγ and of the three IFNγ-induced chemokines CXCL9, CXCL10 and CXCL11 in patients sampled during active sJIA without MAS at sampling divided according to the presence or absence of a history of MAS in their disease course.
Correlations with laboratory abnormalities of MAS
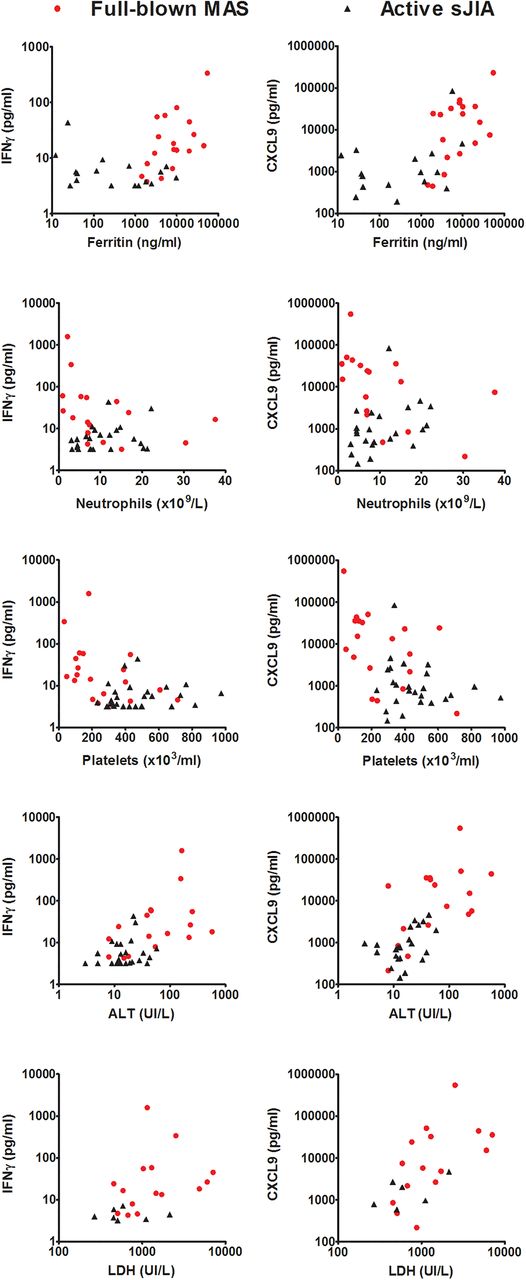
In patients with active sJIA without MAS, levels of IFNγ and of the IFNγ-induced chemokines were not associated with laboratory parameters of MAS with one exception: levels of CXCL9, CXCL10 and CXCL11 were weakly correlated with alanine aminotransferase (ALT) levels with r2 ranging from 0.17 to 0.25 (table 2). The significance of this association is unclear: notably, ALT levels were within normal range in all patients with active sJIA without MAS. In patients with MAS at sampling, no significant correlation of MAS laboratory features with IL-1β and IL-6 was found. In contrast, levels of IFNγ and of the IFNγ-induced chemokines were correlated with ferritin levels, neutrophil and platelet counts, and lactate dehydrogenase and ALT, all typically abnormal during MAS (table 2). These correlations were particularly evident for IFNγ and CXCL9 (table 2 and figure 3). One patient with severe MAS, admitted to the intensive care unit with severe central nervous system involvement, had markedly high levels of IFNγ (336.2 pg/mL), CXCL9 (549 400 pg/mL) and CXCL10 (35 066 pg/mL). Our results show that increased production of IFNγ and of the IFNγ-induced chemokines is a feature of active MAS strongly correlating with the severity of MAS laboratory abnormalities.
Correlation of laboratory parameters of disease activity with levels of interferon γ (IFNγ), CXCL9, CXCL10, CXCL11, interleukin (IL)-6 and IL-1β in patients with macrophage activation syndrome (MAS) and in patients with active systemic juvenile idiopathic arthritis (sJIA) without MAS at sampling

Correlation of levels of interferon γ (IFNγ) and CXCL9 with ferritin levels, neutrophil and platelet count and with lactate dehydrogenase (LDH) and alanine aminotransferase (ALT) levels in patients with active macrophage activation syndrome (MAS) at sampling (red circles) and in patients with active systemic juvenile idiopathic arthritis (sJIA) without MAS at sampling (black triangles). Spearman correlation coefficient (r) and significance level (p) of each correlation are shown in table 2.
Correlation of IFNγ with levels of IFNγ-induced chemokines in patients with MAS
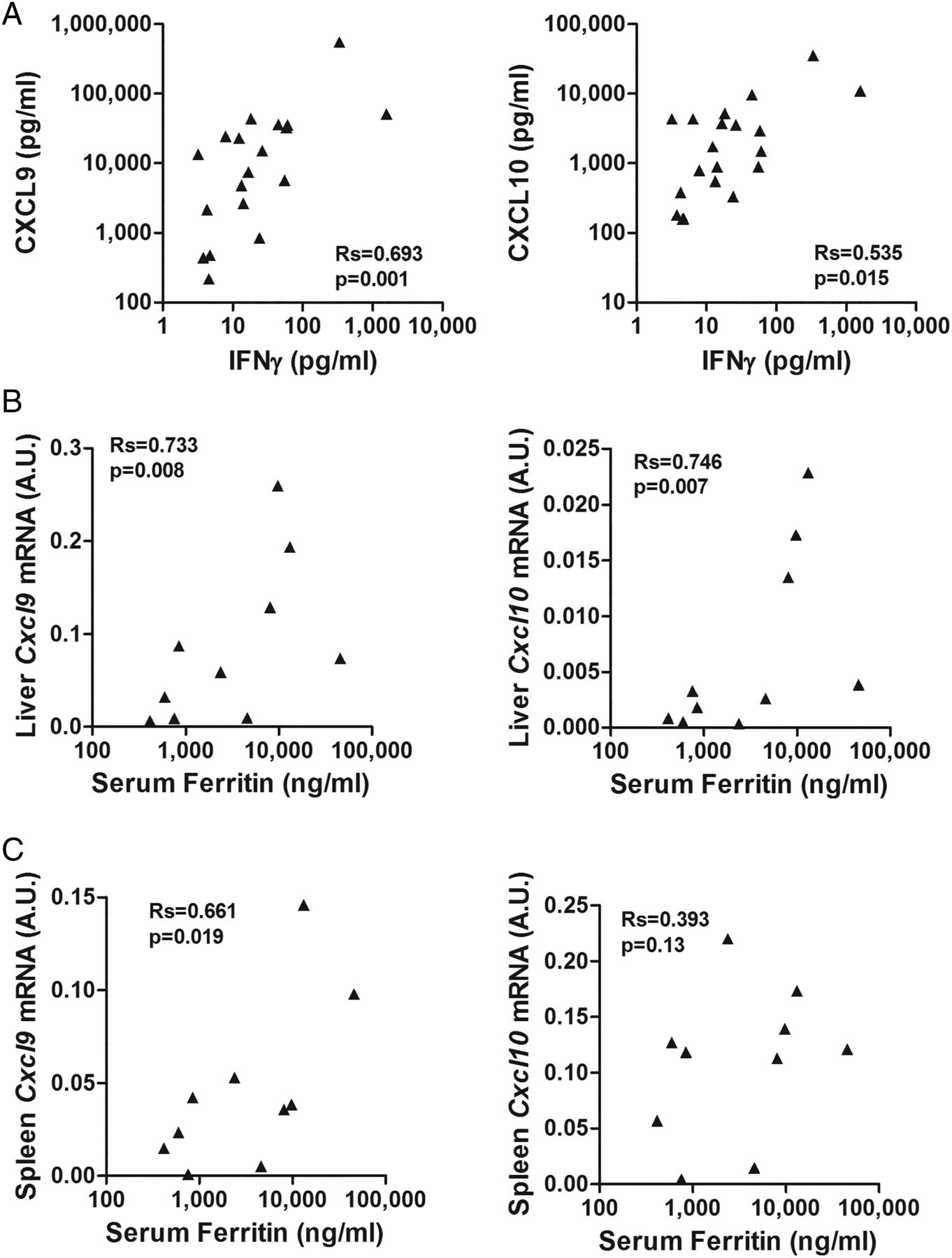
We then evaluated the correlation of IFNγ levels with the levels of each individual chemokine. Notably, CXCL9 appears to be primarily and specifically induced by IFNγ, while CXCL10 and CXCL11 are also induced by type I interferons.22 In patients with MAS, circulating levels of IFNγ were significantly correlated with CXCL9 (r=0.693; r2=0.48; p=0.001), but had a weaker correlation with CXCL10 levels (r=0.535; r2=0.29; p=0.015) (figure 4). The correlation with CXCL11 levels did not reach statistical significance (r=0.447; r2=0.20; p=0.08) (not shown).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Relation of interferon γ (IFNγ) with CXCL9 and CXCL10 production in macrophage activation syndrome (MAS). (A) Correlations of the levels of IFNγ with the levels of CXCL9 and CXCL10 in patients with MAS at sampling. Spearman correlation coefficient (r) and significance level (p) of each correlation are shown. (B, C) Correlation of the levels of Cxcl9 and Cxcl10 mRNA in liver and spleen with serum ferritin levels in murine MAS. In the IL-6TG mouse model of MAS, IFNγ-induced chemokines expression correlates with disease activity. IL-6TG mice were challenged with lipopolysaccharide (5 μg/g body weight) and, 30 hours after the challenge, mRNA expression levels of Cxcl9 and Cxcl10 were assayed in liver (B) and spleen (C) and the results obtained (expressed as arbitrary units) were correlated to serum levels of ferritin at the time of the sacrifice. A base-10 log scale is used for the x-axis of graphs (B, C).
IFNγ-induced chemokines correlate with disease activity in the mouse model of MAS
We investigated the expression of these chemokines in target tissues (liver and spleen) in the murine model of MAS generated in IL-6 transgenic mice. In this model, features of MAS are induced by mimicking an acute infection with administration of LPS on a background of high IL-6 levels,20 recapitulating what occurs in patients with sJIA in whom an infection may trigger MAS. Notably, serum ferritin levels significantly correlated with expression levels of Cxcl9 in spleen and liver and of Cxcl10 in liver (figure 4B and C), demonstrating a correlation of IFNγ-related upstream events in target tissues (ie, liver and spleen Cxcl9 and Cxcl10 expression) and downstream abnormalities such as increased ferritin.
Discussion
Studies in pHLH, in both humans and animals, have demonstrated a central role of IFNγ. However, the role of IFNγ in sec-HLH, including MAS during sJIA, has remained unclear. We show conclusively that high levels of IFNγ and of IFNγ-induced chemokines are present in patients with MAS occurring in sJIA. Additionally, levels of IFNγ, CXCL9 and CXCL10 strongly correlate with laboratory parameters of MAS severity.
In this study, serum levels of IFNγ and of the three IFNγ-induced chemokines were comparable between patients with active sJIA and patients with clinically inactive disease. This result argues against a pathogenic role of IFNγ in sJIA and is, indeed, consistent with a number of published observations. Three gene expression studies failed to find a prominent IFNγ-induced signature in peripheral blood mononuclear cells (PBMCs) of patients with active sJIA without MAS at sampling.23–25 After ex vivo stimulation of PBMCs, the number of cells producing IFNγ in patients with active sJIA was similar to that of controls.26 Consistently, patients with both active and inactive sJIA do not exhibit increased serum or synovial fluid IFNγ levels.27 Supporting the limited role of IFNγ in the arthritis of sJIA, and differently from what found in oligoarticular or polyarticular JIA, CXCL9 and CXCL10 are almost undetectable in the synovial tissues of patients with sJIA.28 Additionally, stimulation of IFNγ knockout mice with Freund's complete adjuvant produces a systemic inflammatory syndrome that includes features of sJIA.29
In contrast, we have shown markedly higher levels of IFNγ and of IFNγ-induced chemokines in patients with MAS at sampling compared with patients with active sJIA without MAS. In patients sampled during MAS, levels of IL-6 or IL-1β were not increased nor associated with laboratory parameters of MAS, suggesting that these cytokines, although critically involved in sJIA pathogenesis,30 ,31 may not be crucial in MAS. It should be noted that, given the well-known difficulties in reliably measuring the low levels of IL-1β in blood, caution should be used in drawing conclusions based on data limited to circulating IL-1β levels.
The mechanisms leading to the marked activation of IFNγ production during MAS on a background of active sJIA remain to be clarified. Identification of patients carrying heterozygous variants of the same genes involved in pHLH,5–9 and the transient defect in cytotoxic NK activity secondary to active inflammation,32 suggests that mechanisms, at least in part, similar to those of pHLH may contribute.33 An additional contribution may be represented by high levels of IL-18 that have been suggested to predispose to MAS during sJIA:34 IL-18 is a well-known IFNγ-inducing cytokine.35 Our finding of elevated levels of IFNγ and of IFNγ-induced chemokines is consistent with previous observations. Levels of neopterin, a catabolite of guanosine triphosphate synthesised by macrophages upon IFNγ stimulation, were shown to be higher in patients with MAS compared with patients with active sJIA without MAS.36 Recently, elevated levels of IFNγ and CXCL10 were reported in five patients with HLH, three of whom had MAS in the course of sJIA. Consistently with our results, five patients with active sJIA without MAS at sampling had markedly lower levels of IFNγ and CXCL10.37
Interestingly, we found that not only were the levels of IFNγ and of the IFNγ-induced chemokines markedly elevated, but also that their levels, particularly those of CXCL9, were strictly correlated with MAS laboratory features, indicating association with disease severity. Consistently, we found the highest levels of IFNγ and of CXCL9 and CXCL10 in the one patient with severe disease, multiple organ failure and central nervous system involvement.
In patients with MAS, of the three IFNγ-induced chemokines, CXCL9 was found to have the strongest correlation with IFNγ levels. This observation is consistent with the established notion that CXCL9 production appears to be induced specifically and only by IFNγ, while CXCL10 and CXCL11 can be induced also by type I interferons.22 Indeed, using a murine model of MAS mimicking the triggering of MAS by an infectious stimulus on a background of high IL-6 levels,20 we found that expression levels of CXCL9 in liver and spleen correlated significantly to circulating ferritin levels. For CXCL10 expression level, this correlation was present only for liver. This is also consistent with our findings in patients with MAS, where CXCL9 levels were strictly correlated with all laboratory parameters of MAS. Taken together, these observations in humans and in mice show that CXCL9 strongly correlates with MAS features and IFNγ production, further supporting the hypothesis that excessive production of IFNγ plays a major role in MAS. Our observations are also consistent with the data using serial lymph-node biopsies from a single patient with sJIA: CXCL10 and indoleamine 2,3-dioxygenase, both IFNγ-inducible proteins, were highly expressed in the tissue obtained during MAS, but not in that obtained during active sJIA without MAS.37 Altogether, these data lead to the hypothesis that circulating levels of CXCL9, and possibly of CXCL10, reflect tissue production of these chemokines in response to local IFNγ production and may therefore serve as a MAS biomarker both in the diagnostic process and in the evaluation of disease activity. This hypothesis is being tested in a larger multicentre study.
Our results in MAS and in sec-HLH, together with the observations available in the literature in patients with pHLH, support the hypothesis that an increase in IFNγ and in IFNγ-induced chemokines, particularly CXCL9, is a characteristic feature of HLH, independently of the underlying cause. In this respect, it is interesting to note that high levels of CXCL9 were reported in the patient with relapsing MAS caused by an NLRC4 gain-of-function mutation.38 We have also observed high levels of CXCL9 in a serum of patients with NLRC4-induced disease (unpublished). This suggests that also in the setting of an HLH induced only by inflammasome dysregulation IFNγ hyperproduction is in place.
Data in animal models of pHLH, both in perforin and in Rab27a knockout mice, unequivocally demonstrate the pathogenic role of IFNγ.10 ,11 Similarly, recent data in the TLR9-induced model of HLH, a model of HLH secondary to infection, have also shown a major role for increased IFNγ production39 (Buatois, 2015, submitted). We have recently demonstrated in the above-mentioned murine model of MAS that treatment with an anti-IFNγ antibody led to increase in survival and reverted clinical and laboratory features of MAS (Prencipe, 2015, submitted). Altogether, these data in different animal models of primary and secondary HLH provide evidence that IFNγ may be the common pivotal mediator of the clinical syndrome, whatever the underlying cause. Encouraging preliminary efficacy and safety data of the phase 2 clinical trial in primary HLH with NI-0501, an anti-IFNγ monoclonal antibody, have been recently reported.40 Our results provide the rationale for IFNγ neutralisation as a therapeutic approach in MAS.
References
Footnotes
Handling editor Tore K Kvien
Contributors CB, CdM, GP, FDB: study hypothesis and design, writing and reviewing manuscript; CB GS, AAG, SG, AR: samples and clinical and laboratory data from patients. CB, KdG, DPM: correlations and statistical analysis. FG and WF performed the multiplex assays. GP and IC: animal experiments. All the authors reviewed, commented, and approved the final manuscript.
Funding AAG is supported by NIH grants R01-AR059049, and P01-AR048929; GS is supported by a Scientist Development Award from the Rheumatology Research Foundation; GP is supported by the Italian Ministry of Health (Rome, Italy) Young Investigator Grants ‘GR-2011-02347874’. This study was realised in the context of an EU-funded FP7 project, named FIGHT-HLH (306124) coordinated by CDM.
Competing interests KdG, FG, WF, CDM: Novimmune; AAG: Novimmune, Novartis Pharmaceutical Corporation, Roche Pharmaceuticals; FDB: Novartis, Novimmune, Hoffmann-La Roche, SOBI, AbbVie.
Parental/guardian consent Obtained.
Ethics approval Ospedale Bambino Gesu’ Ethical Committee, Istituto Giannina Gaslini Ethical Committee, Cincinnati Children's Hospital Ethical Committee.
Provenance and peer review Not commissioned; externally peer reviewed.