Article Text

Abstract
Objective Cardiomyopathy is among the leading causes of death from systemic sclerosis (SSc). Urokinase-type plasminogen activator receptor (uPAR)-deficient mice have been recently reported to display important histopathological hallmarks of SSc, including dermal fibrosis, reduced dermal capillary density, and pulmonary fibrosis. Here, we investigated whether uPAR-deficient mice could display the histopathological features of SSc-related cardiomyopathy.
Methods Ventricular myocardial specimens from uPAR-deficient and wild-type mice at 12 and 24 weeks of age were analysed by both light microscopy and transmission electron microscopy. Picrosirius red staining and hydroxyproline content of myocardial specimens were quantified. Myofibroblast and microvessel counts were determined by immunofluorescence for α-smooth muscle actin and CD31, respectively. Endothelial cell apoptosis was assessed by a combined TUNEL/CD31 immunofluorescence assay. Expression of uPAR in human SSc and control ventricular myocardial autopsy specimens was determined by immunohistochemistry.
Results The myocardium of 24-week-old uPAR-deficient mice displayed focal ischaemic lesions with cardiomyocyte hypertrophy, myofibril rarefaction and contraction band necrosis. At 24 weeks of age, interstitial and perivascular collagen deposition and myofibroblast counts were significantly greater in myocardial tissue of uPAR-deficient mice than in wild-type mice. In uPAR-deficient mice, myocardial fibrosis was paralleled by microvascular endothelial cell apoptosis and reduced capillary density. uPAR expression was significantly downregulated in the myocardium of patients with SSc.
Conclusions Typical histopathological features of SSc-related cardiomyopathy are mimicked by uPAR-deficient mice. The downregulation of uPAR in the myocardium of patients with SSc may suggest similar underlying pathogenetic mechanisms. uPAR-deficient mice could be used as a preclinical model to study the mechanisms and therapeutic approaches of myocardial involvement in SSc.
- Systemic Sclerosis
- Cardiovascular Disease
- Qualitative research
Statistics from Altmetric.com
Introduction
Systemic sclerosis (SSc) is a complex connective tissue disease characterised by widespread microvascular damage and progressive fibrosis involving the skin and multiple internal organs, especially lungs, heart and gastrointestinal tract.1 During the disease course, the progressive fibrotic process disrupts the physiological structure of the affected tissues and commonly leads to significant organ dysfunction.1 Although often clinically occult, cardiac involvement is thought to occur in the majority of patients with SSc accounting for up to one-third of deaths.2–4 Clinical manifestations of primary cardiac involvement are important negative prognostic factors and include arrhythmias, disturbances of the coronary and conduction systems, myocarditis, pericarditis, diastolic as well as systolic ventricular dysfunction, and ultimately congestive heart failure.2 ,5 Furthermore, in SSc, cardiac complications may develop as a secondary phenomenon due to pulmonary arterial hypertension, interstitial lung disease and kidney pathology.2
The general pathogenetic mechanisms reported for SSc, including microvascular alterations leading to tissue ischaemia and hypoxia, collagen accumulation by activated fibroblasts which transdifferentiate into profibrotic myofibroblasts, and complex immune disturbances, are also thought to be involved in the pathogenesis of SSc-related myocardial involvement.2 ,6 Histologically, patchy distribution of myocardial fibrosis and foci of contraction band necrosis, which are thought to mainly arise from ischaemia–reperfusion injury, are considered pathognomonic features of the disease.2 ,7 ,8 Myocarditis has been reported in SSc patients with recent-onset heart disease and occasionally in patients with acute and severe cardiac symptoms.2 ,9 Recently, endothelial cell apoptosis with reduced capillary density, perivascular inflammation, myofibroblast differentiation and accumulation of collagen have been reported in the myocardium of patients with SSc without clinically manifest cardiac involvement.10
In SSc, the high incidence and severity of primary cardiac involvement have stimulated a growing number of epidemiological and imaging studies. However, owing to the paucity of clinical samples and the lack of suitable animal models, the pathogenetic mechanisms underlying SSc-related cardiomyopathy remain mostly unknown. Interestingly, recent experimental evidence indicates that histopathological features of SSc-related cardiomyopathy may be mimicked by murine models of SSc, especially fos-related antigen 2 (Fra-2) transgenic mice.10 In this context, urokinase-type plasminogen activator receptor (uPAR)-deficient mice are a recently described animal model displaying important histopathological hallmarks of SSc, such as dermal fibrosis, dermal endothelial cell apoptosis with reduced capillary density, and progressive pulmonary fibrosis with non-specific interstitial pneumonia-like features.11 ,12
On these premises, the present study was designed to investigate whether uPAR-deficient mice would display the histopathological features of SSc-related cardiomyopathy.
Materials and methods
Materials and methods and any associated references are available in the online supplement.
Results
On the basis of the main histopathological features commonly reported in the myocardium of patients with SSc,2 ,7 ,10 ,13 we systematically assessed left and right ventricular myocardial specimens from our uPAR-deficient mice, focusing on the possible presence of (i) cardiomyocyte damage, (ii) collagen accumulation and α-smooth muscle actin (α-SMA)-positive myofibroblast differentiation, (iii) perivascular inflammation, and (iv) endothelial cell apoptosis with reduced capillary density. Following our previous report on uPAR-deficient mice,11 two different time points were included in the analyses, namely 12 and 24 weeks of age.
To detect possible cardiomyocyte alterations, we analysed mouse ventricular myocardial specimens by both light microscopy and transmission electron microscopy. Analysis of H&E-stained sections did not reveal any obvious signs of cardiomyocyte damage in 12-week-old uPAR-deficient mice compared with wild-type mice (figure 1A). In contrast, under both light and electron microscopy, the myocardium of 24-week-old uPAR-deficient mice displayed focal ischaemic lesions with hypertrophy of cardiomyocytes, severe rarefaction and loss of myofibrils, and foci of contraction band necrosis (figure 1B, C).

Myocardial histopathology in mice deficient for urokinase-type plasminogen activator receptor (uPAR). (A, B) Representative microphotographs of H&E-stained left ventricular myocardium sections from uPAR-deficient mice and wild-type mice at (A) 12 weeks of age (n=7 uPAR-deficient mice, n=7 wild-type mice) and (B) 24 weeks of age (n=7 uPAR-deficient mice, n=6 wild-type mice). Each panel shows myocardial cross-sections on the left side and myocardial longitudinal sections on the right side. (A) No obvious signs of cardiomyocyte damage are evident in 12-week-old uPAR-deficient mice. (B) Focal ischaemic lesions with hypertrophy of cardiomyocytes, severe myofibril rarefaction, and foci of contraction band necrosis (arrows) can be observed in the myocardium of 24-week-old uPAR-deficient mice. Original magnification ×63. Scale bars=50 µm. (C) Representative transmission electron microscopy microphotographs of left ventricular myocardium ultrathin sections from uPAR-deficient mice (n=6) and wild-type mice (n=6) at 24 weeks of age. In the myocardium of uPAR-deficient mice, the loss of myofibrils results in wide empty areas of cardiomyocyte cytoplasm (arrows). Arrowheads indicate contraction band necrosis. Scale bars=2 µm.
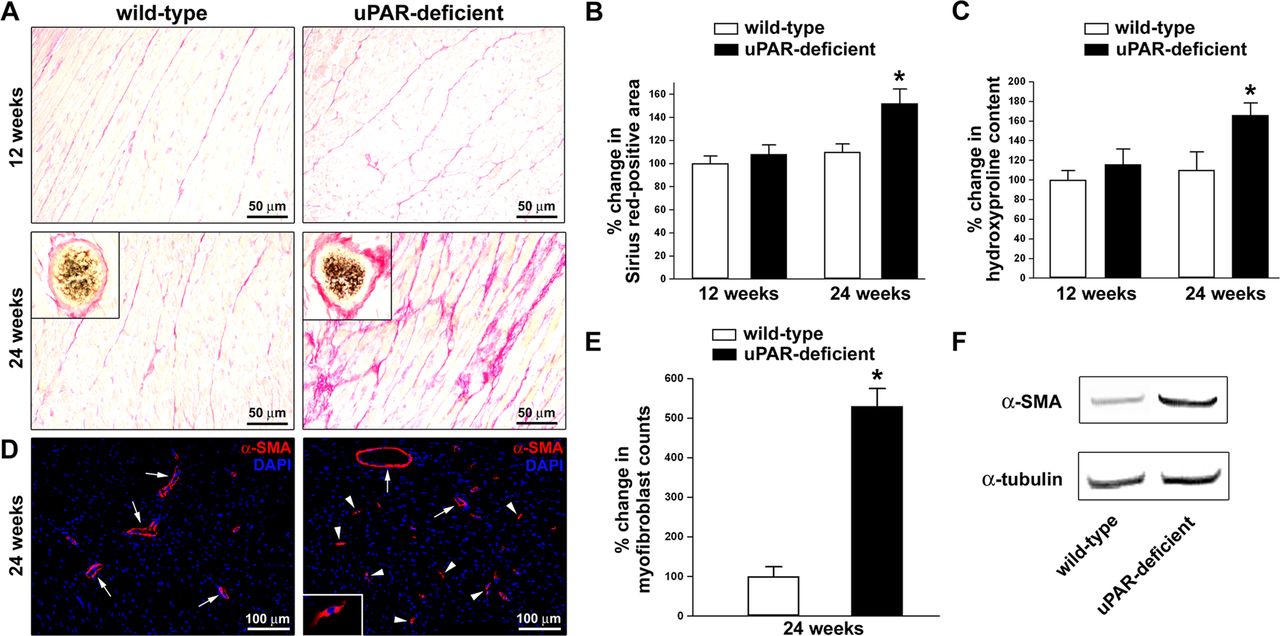
We have recently reported increased deposition of collagen in both the skin and lungs of 12-week-old and 24-week-old uPAR-deficient mice compared with age-matched wild-type littermates.11 Thus, to investigate whether uPAR-deficient mice could even develop myocardial fibrosis, here ventricular myocardium sections from uPAR-deficient and wild-type mice at 12 and 24 weeks of age were stained with Picrosirius red for the visualisation of collagen fibres. As displayed in figure 2A, B, histological examination of Picrosirius red-stained tissue sections demonstrated no difference in left ventricular myocardial collagen deposition between 12-week-old uPAR-deficient mice and wild-type littermates. Conversely, at 24 weeks of age we observed a patchy accumulation of collagen in the left ventricular myocardial interstitium of uPAR-deficient mice (figure 2A). Prominent perivascular fibrosis could also be detected (figure 2A, insets). Indeed, image analysis revealed that the Picrosirius red-positive area was significantly greater in left ventricular myocardium of 24-week-old uPAR-deficient mice than in wild-type littermates (p<0.001) (figure 2B). Consistent with these findings, the hydroxyproline content was significantly increased in left ventricular myocardium specimens from 24-week-old uPAR-deficient mice compared with wild-type mice (p<0.001) (figure 2C). Moreover, in 24-week-old uPAR-deficient mice there was a significant increase in the number of α-SMA-positive profibrotic myofibroblasts than in wild-type mice (p<0.001) (figure 2D, E). Western blotting analysis also revealed a strong increase in α-SMA protein expression in left ventricular myocardial specimens from 24-week-old uPAR-deficient mice compared with wild-type littermates (figure 2F). As shown in online supplementary figure S1A–D, analysis of right ventricular myocardial specimens yielded similar results.

Analysis of collagen content and myofibroblast counts in left ventricular myocardium of mice deficient for urokinase-type plasminogen activator receptor (uPAR) and wild-type mice. (A) Representative microphotographs of myocardial sections from uPAR-deficient and wild-type mice at 12 weeks of age (n=7 uPAR-deficient mice, n=7 wild-type mice) and 24 weeks of age (n=7 uPAR-deficient mice, n=6 wild-type mice) stained with Picrosirius red. Collagen is stained red. No difference in myocardial collagen deposition between 12-week-old uPAR-deficient mice and wild-type littermates can be observed. The myocardium of 24-week-old uPAR-deficient mice displays increased deposition of collagen in the interstitium and in perivascular areas (insets). Original magnification ×40, ×63 (insets). Scale bars=50 µm. (B, C) Quantification of Picrosirius red-positive area (B) and hydroxyproline content (C) both expressed as percentage of that observed in 12-week-old wild-type mice. Data are mean±SD. *p<0.001 versus wild-type mice. (D) Representative microphotographs of left ventricular myocardium sections from 24-week-old uPAR-deficient (n=7) and wild-type (n=6) mice immunostained with rabbit polyclonal α-smooth muscle actin (α-SMA) antibody (red) and counterstained with 4′,6-diamidino-2-phenylindole (DAPI; blue) for nuclei. Myofibroblasts were identified as α-SMA-positive spindle-shaped cells (arrowheads). A higher magnification view of a myofibroblast is shown in the inset. Arrows indicate α-SMA-positive vessels. Original magnification ×20, ×63 (inset). Scale bars=100 µm. (E) Quantification of myofibroblasts in the myocardium of uPAR-deficient and wild-type mice. Data are mean±SD of α-SMA-positive myofibroblast counts expressed as percentage of that observed in wild-type mice. *p<0.001 versus wild-type mice. (F) Western blotting of left ventricular myocardial protein lysates from 24-week-old uPAR-deficient (n=5) and wild-type (n=5) mice analysed using α-SMA antibodies. Representative immunoblots are shown. α-Tubulin antibodies were used as a loading control.
We also assessed mouse myocardial tissues for leucocyte infiltration. However, no relevant signs of perivascular inflammation could be detected in the myocardium of uPAR-deficient mice at either 12 or 24 weeks of age (data not shown).
In 24-week-old uPAR-deficient mice, myocardial fibrosis was paralleled by microvascular endothelial cell apoptosis and reduced capillary density (figure 3A, B). In fact, there was a significant increase in the percentage of apoptotic endothelial cells in parallel with a significant decrease in microvessel counts compared with wild-type littermates (both p<0.001) (figure 3C, D). Similar findings were observed in left and right ventricular myocardium. No differences in endothelial cell apoptosis and microvessel density were found between uPAR-deficient mice and wild-type mice at 12 weeks of age (data not shown).

{kind=link}
{kind=link}
{kind=link}
Increased apoptosis of microvascular endothelial cells and decreased microvessel density in left ventricular myocardium of mice deficient for urokinase-type plasminogen activator receptor (uPAR). (A, B) Representative microphotographs of myocardial sections from 24-week-old uPAR-deficient (n=7) and wild-type (n=6) mice subjected to combined TUNEL (green)/CD31 (red) immunofluorescence assay and counterstained with 4′,6-diamidino-2-phenylindole (DAPI; blue) for nuclei. Apoptotic endothelial cell nuclei are green/blue double stained (arrows). Inset: Higher magnification view of apoptotic endothelial cells. Original magnification ×40, ×63 (inset). Scale bars=50 µm. (C) Semiquantitative analysis revealed a significantly higher percentage of TUNEL-positive endothelial cells in the myocardium of uPAR-deficient mice compared with wild-type mice. Data are mean±SD. *p<0.001 versus wild-type mice. (D) Quantification of CD31-positive vessels in the myocardium of uPAR-deficient and wild-type mice. Data are mean±SD of CD31-positive vessel counts expressed as percentage of that observed in wild-type mice. *p<0.001 versus wild-type mice.
As previously demonstrated in the setting of dermal fibrosis,11 for uPAR-deficient mice to be proposed as an animal model for SSc-related cardiomyopathy, uPAR should also be downregulated in human SSc myocardium. Therefore, we used immunohistochemistry and immunofluorescence to analyse expression of the native full-length form of uPAR in paraffin-embedded ventricular myocardial autopsy specimens from patients with SSc and healthy subjects (see online supplementary figure S2A, B). uPAR immunostaining was significantly decreased in the stromal compartment of SSc myocardium compared with controls, especially in microvessels and fibroblasts (p<0.001) (see online supplementary figure S2A–C).
Discussion
The present study extends our recent findings on uPAR-deficient mice, which have been reported to develop important histopathological hallmarks of SSc, including dermal fibrosis, dermal microvascular endothelial cell apoptosis with reduced capillary density, and interstitial lung disease resembling the non-specific interstitial pneumonia pattern of human SSc.11
Given the high frequency and prognostic relevance of cardiomyopathy in patients with SSc,2–6 together with our limited knowledge of the underlying pathogenetic mechanisms and the lack of suitable preclinical tools, it is of major importance to identify potential mouse models that resemble SSc-related myocardial involvement. In this context, Venalis et al10 have recently investigated myocardial involvement in different murine models of SSc, including Fra-2 transgenic mice, mice with sclerodermatous chronic graft-versus-host disease (GVHD) and TSK-1 mice. Mice with chronic GVHD and TSK-1 mice presented only certain features of SSc-related cardiomyopathy, while Fra-2 transgenic mice, a model with coexisting vascular alterations and multiorgan fibrosis, mimicked all major histopathological features of SSc-associated myocardial disease.10
Here, we show for the first time that uPAR-deficient mice exhibit typical histopathological features of SSc-related cardiomyopathy, including foci of cardiomyocyte damage with contraction band necrosis, endothelial cell apoptosis with reduced capillary density, profibrotic myofibroblast differentiation, and patchy collagen accumulation in interstitial and perivascular areas of the myocardium. Interestingly, we observed the aforementioned features in both left and right ventricular myocardium of 24-week-old uPAR-deficient mice, but not in younger mice (12 weeks of age), as compared with age-matched wild-type littermates. Thus, in these mice, development of myocardial alterations seems to be delayed with respect to skin and lung involvement.11 We have previously shown that, in uPAR-deficient mice, patchy pulmonary fibrosis worsens significantly from 12 to 24 weeks of age,11 suggesting that, in this model, development of SSc-like histopathological features may even be aging-related. Of note, it has been demonstrated that plasminogen is necessary for urokinase-type plasminogen activator (uPA)-induced cardiac fibrosis, but uPAR is not.14 Our present findings indicate that the absence of uPAR is even associated with the accumulation of profibrotic myofibroblasts and the development of interstitial and perivascular myocardial fibrosis in vivo. These results are consistent with those reported in the skin and lungs of uPAR-deficient mice.11 ,12 Moreover, previous in vitro studies have demonstrated that uPAR cleavage/inactivation in fibroblasts is necessary for transforming growth factor-β-induced transition to α-SMA-expressing myofibroblasts, and that downregulation of uPA/uPAR results in increased cell surface integrin expression and adhesion to the extracellular matrix required for the assembly of α-SMA into stress fibres, thus promoting a persistent myofibroblast phenotype.15 ,16 This evidence also strengthens the hypothesis that, in uPAR-deficient mice, tissue fibrosis mainly arises from fibroblast-intrinsic reprogramming, rather than other mechanisms such as inflammation. This might be consistent with the lack of any relevant signs of inflammation observed in the myocardium of our mice, which is, however, somewhat different from SSc-related myocardial disease. Functional in vitro studies on uPAR-deficient murine fibroblasts are ongoing to shed light on the possible molecular mechanisms involved.
In the myocardium of our uPAR-deficient mice, the appearance of interstitial and perivascular fibrosis was paralleled by endothelial cell apoptosis and microvessel rarefaction, as previously described in the dermis.11 Consistent with our findings, uPA was shown to mediate endothelial cell survival via its antiapoptotic effects, which are dependent on the presence of uPAR.17 In addition, the evidence of reduced capillary density in the myocardium of uPAR-deficient mice is in agreement with our previous observations that uPAR cleavage/inactivation has a major role in the impaired angiogenic function of SSc microvascular endothelial cells.18 ,19 Besides histopathological similarities between human SSc and uPAR-deficient mice, the downregulation of uPAR constitutively observed in the myocardium of patients with SSc further suggests the likelihood of shared pathophysiological mechanisms.
Although these data suggest that uPAR-deficient mice might be a preclinical model for the study of the mechanisms and therapeutic approaches of myocardial involvement in SSc, we should consider that our study has some limitations. Functional assessment (eg, by echocardiography and myocardial perfusion scan) would disclose whether myocardial histopathological changes may be associated with arrhythmias and impaired left and right ventricular ejection fraction and/or cardiac index in uPAR-deficient mice. Unfortunately, we were unable to perform such functional assessments. Moreover, our study focused on analyses of left and right ventricles, which have been reported to be most commonly affected in patients with SSc. However, it would also be interesting to investigate possible histopathological changes in other areas of the heart.
In summary, the myocardial changes detected in uPAR-deficient mice mimic important features of SSc-related cardiomyopathy. Taking the high frequency and recognised clinical relevance of cardiac involvement in patients with SSc together with our limited knowledge of the underlying disease mechanisms, our findings raise the possibility that uPAR-deficient mice might serve in the future as a tool to study the pathophysiology of SSc-associated myocardial disease and to evaluate potential therapeutic approaches.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figures
Footnotes
Handling editor Tore K Kvien
LI-M and MM-C contributed equally.
Contributors Study conception and design: MM, IR, LI-M and MM-C. Acquisition of data: MM, IR, MF, SG, PC, LI-M and MM-C. Interpretation of data: MM, IR, LI-M and MM-C. Manuscript preparation: MM, IR, LI-M and MM-C.
Funding Supported by grants from the University of Florence (Progetti di Ricerca di Ateneo to LI-M and MM-C). The work of PC is funded by Long-term Structural Funding Methusalem by the Flemish Government.
Competing interests None declared.
Patient consent Obtained.
Ethics approval The study was approved by the local institutional review board.
Provenance and peer review Not commissioned; externally peer reviewed.