Article Text

Abstract
The objective of this work was to develop and validate a set of clinical criteria for the classification of patients affected by periodic fevers. Patients with inherited periodic fevers (familial Mediterranean fever (FMF); mevalonate kinase deficiency (MKD); tumour necrosis factor receptor-associated periodic fever syndrome (TRAPS); cryopyrin-associated periodic syndromes (CAPS)) enrolled in the Eurofever Registry up until March 2013 were evaluated. Patients with periodic fever, aphthosis, pharyngitis and adenitis (PFAPA) syndrome were used as negative controls. For each genetic disease, patients were considered to be ‘gold standard’ on the basis of the presence of a confirmatory genetic analysis. Clinical criteria were formulated on the basis of univariate and multivariate analysis in an initial group of patients (training set) and validated in an independent set of patients (validation set). A total of 1215 consecutive patients with periodic fevers were identified, and 518 gold standard patients (291 FMF, 74 MKD, 86 TRAPS, 67 CAPS) and 199 patients with PFAPA as disease controls were evaluated. The univariate and multivariate analyses identified a number of clinical variables that correlated independently with each disease, and four provisional classification scores were created. Cut-off values of the classification scores were chosen using receiver operating characteristic curve analysis as those giving the highest sensitivity and specificity. The classification scores were then tested in an independent set of patients (validation set) with an area under the curve of 0.98 for FMF, 0.95 for TRAPS, 0.96 for MKD, and 0.99 for CAPS. In conclusion, evidence-based provisional clinical criteria with high sensitivity and specificity for the clinical classification of patients with inherited periodic fevers have been developed.
- Familial Mediterranean Fever
- Fever Syndromes
- Inflammation
Statistics from Altmetric.com
Introduction
Autoinflammatory diseases include monogenic and multifactorial inflammatory conditions characterised by exaggerated activation of innate immunity in response to exogenous or endogenous stimuli, in the absence of high-titre autoantibodies.1 Most of these disorders are characterised by recurrent episodes of fever and are defined as periodic fevers. Familial Mediterranean fever (FMF) is an autosomal recessive (AR) disease secondary to mutations of the MEFV (MEditerranean FeVer) gene.2 ,3 It is characterised by short episodes of fever (24–72 h) associated with serositis and arthralgia/arthritis. Mevalonate kinase deficiency (MKD; an AR disease) is caused by loss of function of mevalonate kinase (MVK), an enzyme involved in cholesterol biosynthesis.4 ,5 A partial enzymatic defect causes episodes of fever lasting 4–6 days associated with abdominal pain, diarrhoea, rash and lymph node enlargement.6 The almost complete absence of enzymatic activity is responsible for a severe metabolic disease (mevalonic aciduria) with chronic inflammation and severe neurological impairment. Tumour necrosis factor (TNF) receptor-associated periodic fever syndrome (TRAPS) is an autosomal dominant (AD) disease secondary to mutations of type 1 TNF receptor (TNFSRF1A).7 Fever episodes last more than 6 days and are associated with myalgia, rash and abdominal pain.8 Cryopyrin-associated periodic syndromes (CAPS) are a group of disorders associated with heterozygous mutations of NLRP3, encoding cryopyrin.9 The clinical spectrum of CAPS is broad, ranging from a severe chronic infantile multisystemic inflammatory disease, defined as chronic infantile cutaneous neurological articular (CINCA) syndrome (or neonatal-onset multisystemic, chronic inflammation disease (NOMID)), to a milder phenotype with recurrent episodes of fever, urticarial rash and arthralgia/arthritis.10
Inherited periodic fevers have been observed in all studied ethnicities and populations, although FMF has a particularly high prevalence in Turkish, Arab, Armenian and non-Ashkenazi Jewish populations.11 Disease onset is usually in the first years of life. However, a variable proportion of patients (especially those with FMF and TRAPS) might present first symptoms in their second or third decade of life.11 ,12 Typical ‘inflammatory’ fever episodes can also be observed in a relatively common non-monogenic autoinflammatory disease, named PFAPA (periodic fever, aphthosis, pharyngitis and adenitis) syndrome, characterised by strikingly regular episodes of fever variably associated with at least one of the three manifestations in the acronym in the absence of signs of infection.13
The diagnosis of inherited periodic fevers relies on careful interpretation of the clinical phenotype and results from molecular genetic analysis. Molecular analysis is able to provide a definitive diagnosis in most patients, but the results can be inconclusive or even misleading in other cases.14 As a result, there have been previous attempts to provide clinical guidelines and diagnostic flowcharts to identify appropriate cases for testing.6 ,15–17
Formal diagnostic criteria have been developed for some inherited periodic fevers (FMF and mild CAPS) based on the main clinical manifestations associated with the specific disease within the context of limited populations, and there is some question of their suitability for use in other populations.18–21
The aim of the present study was to take advantage of a large international registry of autoinflammatory diseases (Eurofever) to develop and validate evidence-based clinical classification criteria for the four main autoinflammatory periodic fevers in children and adults.
Patients and methods
Data were extracted from the Eurofever Registry.11 The main characteristics of the registry, the diseases involved and the method of selecting the variables included in the forms have already been described11 ,22 (see online supplementary appendix I). Ethics committee approval for entering patients in the registry and informed consent or assent were obtained in the participating centres, depending on each country's regulations. For the purpose of this study, the following diseases characterised by periodic/recurrent fever episodes were analysed: FMF, MKD, TRAPS and CAPS. Patients with PFAPA were used as disease controls.
Selection of the ‘gold standard’ group and statistical analysis
The Eurofever Registry Steering Committee has appointed a group of experienced clinicians (SO, HO for FMF; JF, AS for MKD; HL, MG, PW for TRAPS; BN, JK-D for CAPS; MH, MG for PFAPA) to evaluate web-collected cases available in the registry. The disease experts have the mandate to control the consistency and quality of the data. In the case of inconsistency or other uncertainty, specific queries are resubmitted to the participating centres for resolution.
The reference ‘gold standard’ group includes patients with FMF, TRAPS, CAPS or MKD with a confirmatory molecular analysis14 defined as follows:
-
FMF: two MEFV mutations, of which at least one is in exon 1023;
-
MKD: two MVK mutations with the exclusion of variants with an uncertain pathological role (such as S52N P165L, H20Q) (http://fmf.igh.cnrs.fr/infevers/)23;
-
TRAPS: heterozygous TNFRSF1A mutations with the exclusion of low-penetrance (such as R92Q or P46L) or uncertain mutations (http://fmf.igh.cnrs.fr/infevers/)23;
-
CAPS: heterozygous NLRP3 mutations with the exclusion of low-penetrance variants (V198M), functional polymorphisms (Q703K) or variants with uncertain pathological role (http://fmf.igh.cnrs.fr/infevers/).23
Other patients with a non-confirmatory genetic test (eg, one mutation in AR disease, low-penetrance mutations, polymorphisms) were considered to be ‘genetically uncertain patients’ and were excluded from the statistical analysis. With the exclusion of patients with severe CAPS presenting a neonatal-onset chronic disease course, the majority of patients with a confirmatory genetic test showed a recurrent disease course (see below). For this reason, patients with a chronic disease course were not considered for the elaboration of the criteria. Patients with PFAPA were classified according to current diagnostic criteria.24 Before the analysis, the centres were retrospectively contacted and asked whether, during the follow-up after enrolment, the diagnosis of PFAPA could be confirmed or if a different diagnosis was pointed to. Patients whose disease was not confirmed by the centres or who were lost to follow-up were excluded. So that the classification criteria could be developed and subsequently validated on an independent set of patients, the gold standard group was randomly split into two subgroups in a ratio of 3:2. The first (‘training set’) was used to identify clinical variables that were able to correctly classify each disease through a classification score. The second group (‘validation set’) was used to verify the performance of the classification score created on the training set.
Statistical analysis was performed and clinical criteria were formulated on the basis of a univariate and multivariate analysis of the training set and validated on the validation set, as previously described16 (see online supplementary appendix II).
Results
Selection and characterisation of the gold standard group
From November 2009 to March 2013, 2556 patients (1258 male, 1298 female) were collected in the Eurofever Registry by 91 centres in 56 countries (see online supplementary figure S1). Of these 2556 patients, 658 were excluded because they had not yet been checked by experts; 590 with confirmed autoinflammatory disease not associated with periodic fever (deficiency of IL-1 receptor agonist (DIRA), pyogenic arthritis, pyoderma gangrenosum and acne (PAPA), chronic recurrent multifocal osteomyelitis (CRMO), Blau's syndrome) and 93 with a chronic disease course (58 CAPS, 13 FMF, 14 TRAPS, 8 MKD; see online supplementary figure S2) were also excluded. The remaining 1215 patients with periodic fevers (498 FMF, 112 MKD, 164 TRAPS, 105 CAPS, 336 PFAPA) were evaluated. A total of 518 patients with inherited periodic fevers were selected as the gold standard group (291 FMF, 74 MKD, 86 TRAPS, 67 CAPS). The other 361 patients with inherited periodic fevers were classified as genetically uncertain patients. In addition, 199 patients with PFAPA were included in the study as disease controls, after final confirmation by the centres at the last follow-up (see online supplementary figure S2).
The main demographic and clinical features of the gold standard patients and patients with PFAPA are reported in table 1. The results of the molecular analysis are reported in online supplementary table S1. At the time of enrolment, 483 (67.3%) patients were paediatric (<14 years) and 234 (33.7%) were adults. Disease onset was reported during childhood in 671 patients (93.7%) (see online supplementary figure S3).
Principal demographic features and clinical manifestations in gold standard patients
Development of a clinical classification score and performance in the validation set
The 518 gold standard and 199 PFAPA patients were randomly split into a training (n=412) and a validation (n=305) set; the main demographic characteristics of the two groups are summarised in online supplementary table S2. Univariate analysis performed on the training set identified clinical variables associated with each disease (see online supplementary table S3). The results of multivariate analysis performed on the training set are reported in table 2. For each disease, the symptoms that independently discriminate it from the other disorders are reported, together with the weights estimated by the logistic model. The score for each disease is calculated by summing all the weights associated with the presence or absence of symptoms in each patient (table 3).
Results of the multivariate analysis in gold standard patients (training set)
The Eurofever clinical diagnostic/classification criteria*
The discriminative ability of the linear scores calculated for each disease was assessed on the training set by receiver operating characteristic (ROC) curve analysis (figure 1); for each disease, an optimal cut-off, based on the point on the ROC curve giving maximum accuracy, was chosen to classify patients as diseased/not diseased. The scores and cut-offs calculated on the training set according to the above procedure were then applied to the validation set, calculating the sensitivity and specificity of the score on this independent set of patients. In figure 1 the performance of the classification criteria on the training set is compared with the performance obtained with the validation set. All criteria displayed high sensitivity and specificity, with an area under the curve above 0.90 in all subgroups (figure 1).

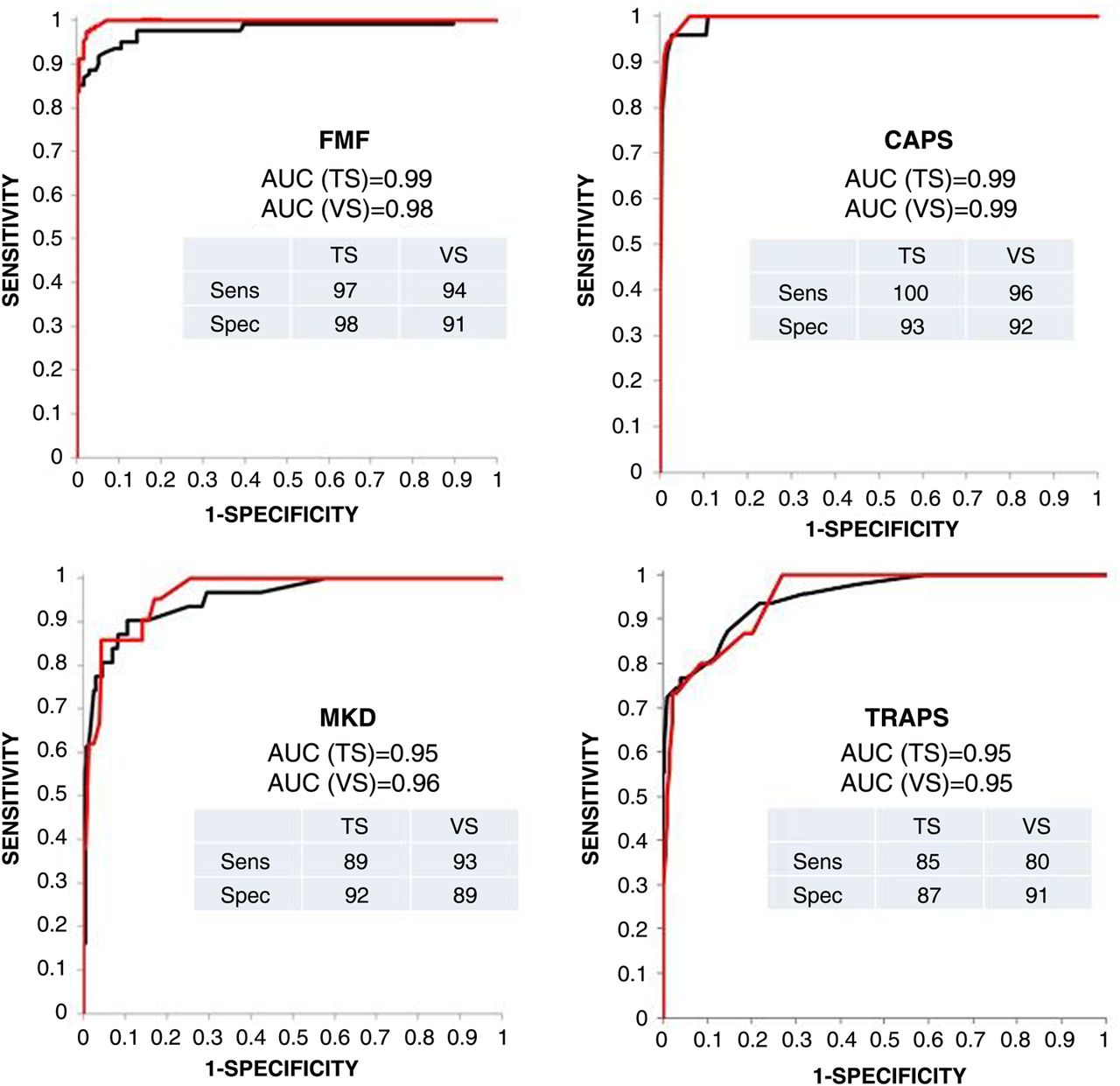{kind=link}
Receiver operating characteristic curves obtained for training (TS) and validation (VS) sets of gold standard patients, and the sensitivity (Sens) and specificity (Spec) of each classification criterion. AUC, area under the curve; CAPS, cryopyrin-associated periodic syndromes; FMF, familial Mediterranean fever; MKD, mevalonate kinase deficiency; TRAPS, receptor-associated periodic fever syndrome.
The performances of the four scores providing the best accuracy in the total group of gold standard patients (validation and training sets) according to the different diseases are shown in online supplementary figure S4. In 144 patients (19.7%), a double classification was obtained. In this case, the threshold of increase above the cut-off value related to the correct diagnosis was generally higher than those obtained for the incorrect diagnosis (see online supplementary table S4). Only 10 patients (1.3%) did not receive any classification.
Different cut-off values providing a higher sensitivity and the cut-off values providing a higher specificity for each disease are also shown in online supplementary figure S4. Even with a lower specificity (see legend to online supplementary figure S4), the use of a ‘high-sensitivity score’ would allow the identification of more than 95% of patients, minimising the risk of excluding possibly affected patients from the molecular analysis during the diagnostic work-out.
Performance of the classification score for patients with a non-confirmatory genetic test (genetically uncertain patients) and patients with a chronic disease course
The clinical and molecular features of 361 patients without a confirmatory genetic test are reported in online supplementary tables S5 and S6, and performances of the classification criteria in this subgroup are reported in table 4. The overall specificity of the most accurate criteria was generally high. The highest numbers of FMF-like patients who were positive according to the FMF score were those carrying two MEFV mutations not in exon 10, the heterozygous patients with mutations in exon 10, and patients not genetically screened (75%). A similar percentage of positivity was observed in CAPS-like patients, including those carrying the V198M low-penetrance variant and the Q703K polymorphism. Conversely, only 52% of patients carrying the R92Q variant of TNFRFS1A, a low-penetrance mutation, usually associated with a milder phenotype,25 were positive according to the score.
Performance of Eurofever classification criteria in genetically uncertain patients
We also verified the performance of classification criteria in the group of patients with a chronic disease course (see online supplementary table S7). The vast majority of CAPS patients with a chronic disease course (mainly CINCA/NOMID) were positive according to the Eurofever classification criteria. The same was observed for patients with other diseases, especially those with a confirmatory genetic test (see online supplementary table S8).
Discussion
We propose a new set of provisional classification criteria for patients with inherited autoinflammatory diseases presenting with periodic fever. Multivariate analysis on a large group of patients with different periodic fevers has allowed identification of a set of variables that gave a very high performance in an independent group of patients. These criteria are aimed to help experts in the field correctly clinically classify patients with suspected autoinflammatory disease and should be applied only after careful exclusion of other causes of periodic fevers, such as infections, immunodeficiency, neoplasms, and other rheumatic conditions with uncertain genotype.
A factual limitation of the study was the decision to create the criteria on the basis of clinical findings observed in gold standard patients with a confirmatory genetic test. This approach potentially overemphasises ‘classical’ presentations of the diseases, limiting recognition of patients with atypical phenotypes. Certainly, clinical criteria need to be considered in the light of information from molecular analysis, and vice versa, they need to enable recognition of patients with clear-cut pathogenic mutations even with an unusual clinical presentation. For this reason, we propose to attribute the term ‘provisional’ to the proposed criteria.
All diagnostic or classification criteria and guidelines for genetic analysis available in the literature to date have been developed on the basis of expert opinion or on evaluation of clinical manifestations in patients affected by a single disease, usually in the context of a limited population or ethnic background.6 ,8 ,15–20 ,24 The wide overlap of the clinical features associated with episodes of fever in these conditions is the major cause of the low performance of these diagnostic criteria when applied to patients affected by different autoinflammatory diseases.16 ,26 In the present study, we followed an alternative approach to the previous classical consensus of experts, which is commonly used for diseases for which a specific diagnostic marker is lacking.27 The availability of the large international Eurofever Registry has, for the first time, enabled comparison of patients with different diseases, but with a common data collection, and of heterogeneous geographic and ethnic distribution. This approach allowed identification of ‘positive’ and ‘negative’ criteria correlated with each disease, resulting in the high accuracy observed for each set of criteria.
This new set of criteria might represent a useful practical tool to be used in daily clinical practice for patients with suspected autoinflammatory disease—either for the selection of genes suitable for molecular analysis and for their final classification after genetic tests, or when an unpublished genetic mutation is found in a given patient, or when the genetic testing is not clearly confirmatory. In the first case, the use of the ‘high-sensitivity score’ would minimise the risk of excluding possible positive patients from genetic analysis.
Depending on the pattern of inheritance, the identification of one or two mutations with known pathogenic impact and high penetrance represents an essential final step in the diagnosis of monogenic autoinflammatory diseases.14 However, in a considerable proportion, molecular analysis is unable to provide diagnostic confirmation—for example, in the case of a single mutation in AR disorders or the identification of variants of unknown significance such as low-penetrance mutations, functional polymorphisms, and novel variants of unknown functional impact.14 ,28 To further complicate this issue, the extensive use of molecular analysis over the last few years has revealed a growing number of patients carrying mutations in more than one gene.29 Non-confirmatory genetic results provide a challenge for both physicians and geneticists and may lead to overestimation of the pathogenic relevance of genetic variants in patients presenting with an unclear inflammatory phenotype.14 This problem will become more pressing with the application of next-generation sequencing, a technique that holds promise as a potent diagnostic tool for periodic fevers and other genetic disorders. This will almost certainly result in identification of a huge number of variants of unknown significance in the genes associated with periodic fevers. As a result, studies such as this, which both correlate and validate data from molecular analysis and the clinical phenotype, will become more critical both for correct classification of patients and assessing the impact or otherwise of genetic variants.
Application of the Eurofever classification criteria in patients without genetic confirmation and in patients with a chronic disease course revealed some interesting features. Despite some variability related to the different genotypes, a high proportion of patients with a non-confirmatory genetic test in the present study turned out to be positive with a high accuracy score. These results should nonetheless be interpreted with caution, as it is probable that the considerable number of these patients fulfilling the clinical classification criteria in this study is due to a bias in patient selection by the registry, which is strongly predisposed towards patients for whom the enrolling centres have a serious suspicion of a given disease.12 It is likely that application of the new classification criteria in daily practice, in which a less rigorously selected population is present (patients with a non-confirmatory genetic test or positive for more than one gene, undifferentiated patients with a negative genetic test), might influence the actual accuracy of the present criteria. This possibility is being verified in a prospective validation of the criteria in a random population of patients with suspected autoinflammatory diseases.
Even though the criteria were developed and validated in patients with periodic fever, the performance of the diagnostic/classification criteria was also particularly high in patients presenting with a chronic disease course. Even though 97% of CAPS patients with a chronic disease course were correctly identified by the present criteria, it is conceivable that CAPS merits specific diagnostic/classification criteria that could cover all possible NLRP3-associated phenotypes, including those clinical features (severe neurological involvement, bone alterations, etc) related to the most severe clinical phenotype (CINCA/NOMID), usually presenting with a chronic inflammatory disease course from birth. For similar reasons, we believe that the present score is not suitable for the diagnosis and classification of the most severe form of MKD deficiency, mevalonic aciduria.
In conclusion, we present a validated evidence-based tool either for indication for molecular analysis or for clinical classification of patients with suspected autoinflammatory periodic fevers after careful exclusion of other causes. Future work building on this will include prospective validation of the criteria in everyday clinical practice and a consensus process among paediatric and adult clinicians and genetic experts in the field to generate guidelines for the correct combination of these clinical classification criteria and other possible clinical variables, such as response to treatment or specific laboratory examinations (eg, urinary mevalonic acid for MKD), with information from molecular analysis to provide definitive classification of patients with autoinflammatory periodic fevers.
Acknowledgments
We thank the research assistants, Eugenia Mosci and Irene Gregorini, for their valuable and excellent work. We also thank all members of Eurofever/PRINTO who participated as investigators in the study on patients with periodic fevers and whose enthusiastic efforts made this work possible: Argentina: Graciela Espada, Ricardo Russo, Carmen De Cunto; Australia: Christina Boros; Chile: Arturo Borzutzky; Croatia: Marija Jelusic-Drazic; Czech Republic: Pavla Dolezalova; Denmark: Susan Nielsen; France: Veronique Hentgen; Germany: Tobias Schwarz, Rainer Berendes, Annette Jansson, Gerd Horneff; Greece: Efimia Papadopoulou-Alataki, Elena Tsitsami, Florence Kanakoudi Tsakalidou; Italy: Romina Gallizzi, Laura Obici, Patrizia Barone, Rolando Cimaz, Mariolina Alessio, Japan: Ryuta Nishikomori; Latvia: Valda Stanevicha; Netherlands: Esther Hoppenreijs; Poland: Beata Wolska-Kusnierz; Romania: Nicolae Iagaru; Russia Federation: Irina Nikishina; Saudi Arabia: Sulaiman M. Al-Mayouf, Sewairi; Serbia: Gordana Susic. Slovenia: Natasa Toplak; Spain: Consuelo Modesto, Maria Jesus Rua Elorduy, Jordi Anton, Rosa Bou.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online Appendix I
- Data supplement 3 - Online Appendix II
Footnotes
Handling editor Tore K Kvien
Contributors SF, MPS, NR and MG: coordination of the study, analysis of the data, manuscript preparation. SO, HJL, GA, PW, IK-P, ND, LC, AI, YU, DR, PQ, ED, TH, AM, GF, TK, SM, AYB, AO, JK-D, BN, AS, HO, IT, JF, MH, AM: data collection and analysis, revision of the manuscript.
-
Funding The Eurofever Registry was sponsored by the Autoinflammatory Diseases’ Working Group of the Paediatric Rheumatology European Society (PRES) and supported by the Executive Agency For Health and Consumers (EAHC, Project No 2007332) and by Coordination Theme 1 (Health) of the European Community's FP7 (Eurotraps, grant agreement number HEALTH-F2-2008-200923). Novartis and SOBI granted unrestricted educational grants.
-
Competing interests None.
-
Ethics approval G Gaslini ethics board.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement Additional material is published online only.