Article Text

Abstract
OBJECTIVE To investigate the existence of differences among European referral centres for systemic sclerosis (SSc) in the pattern of attendance and referral and in the clinical and therapeutical approaches.
METHODS In 1995 the European Scleroderma Study Group initiated a multicentre prospective one year study whose aim was to define the disease activity criteria in SSc. During the study period each participating European centre was asked to enrol consecutive patients satisfying American College of Rheumatology criteria for SSc and to fill out for each of them a standardised clinical chart. Patients from various centres were compared and differences in epidemiological, clinical, and therapeutical aspects were analysed.
RESULTS Nineteen different medical research centres consecutively recruited 290 patients. The patients could be divided into two subgroups: 173 with the limited (lSSc) and 117 with the diffuse (dSSc) form of the disease. The clinical and serological findings for the series of 290 patients seemed to be similar to data previously reported. However, when the data were analysed to elicit any differences between the participating centres, a high degree of variability emerged, in both epidemiological and clinical features and in the diagnostic and therapeutic approaches to the disease.
CONCLUSIONS The clinical approach to SSc, not only in different countries but also in different centres within the same country, is not yet standardised. To overcome this problem, it will be necessary for the scientific community to draw up a standardised procedure for the management of patients with SSc. This would provide a common research tool for different centres engaged in research on this complex disease.
- systemic sclerosis
- clinical differences
- attendance
- referral
Statistics from Altmetric.com
Systemic sclerosis (SSc) is a multisystem connective tissue disorder characterised by widespread microvascular and macrovascular damage, and by fibrosis of the skin and internal organs, particularly the gut, lung, heart, and kidney.1-6 The extent of skin and internal organ involvement, and the severity and course of the disease may vary greatly.7-11 Moreover, the treatment for patients with SSc has not yet been standardised.12 Thus the clinical approach may differ widely not only between doctors at outpatient clinics and those working in research oriented tertiary centres but also between centres of the same type.13-15
In 1995 the European Scleroderma Study Group was formed and began a multicentre study whose aim was to define a valid set of criteria for disease activity in SSc. Data were gathered on a large number of patients consecutively recruited at 19 different medical research centres in Europe. In this paper we report and discuss the preliminary data gathered at the first observation on 290 patients. In addition, by examining groups of consecutive patients from the various centres, we were able to evaluate their patterns of attendance and referral.
Materials and methods
Nineteen centres located in 11 different European countries participated in this study. Each investigator was asked to enrol a consecutive number of patients with SSc, satisfying the preliminary American College of Rheumatology (ACR, formerly American Rheumatism Association) criteria for the classification of the disease.16
CLINICAL CHART
A standardised clinical chart was prepared and one copy was sent to each participating centre. Table 1 summarises the demographic and epidemiological information, and table 2 the clinical, laboratory, and other parameters (radiological, etc) that were to be gathered and recorded for each patient.
Demographic and patient history registered in the clinical chart
Main clinical, laboratory, and other diagnostic parameters investigated
The chart was made up of four sections. In section I, for each patient the participants were asked to provide demographic information and a patient history; an evaluation based on the preliminary criteria for SSc of the ACR; a classification of the patient's disease as diffuse or limited (dSSc or lSSc) according to Le Roy et al.8
At entry, each patient also had to undergo a complete evaluation, and the results (including symptoms, signs, laboratory and other diagnostic test findings) were to be recorded in section II. This section was divided into 11 parts, 10 focusing on specific categories of disease manifestations (generalised complaints; cutaneous manifestations; vascular manifestations; cardiopulmonary manifestations; articular/muscular manifestations; ocular manifestations; gastrointestinal manifestations; haematological alterations; renal alterations; and neuropsychiatric manifestations), and the 11th on aspecific inflammation and immunological features. At the end of each of the 11 subsections, an additional item was included (Δ-factor) designed to note any worsening in the relevant manifestation during the month preceding enrolment (according to the patient's report). Sections III and IV were analogous to section II (except that the Δ-factor recorded any change in the manifestation compared with the previous observation) and were to be completed for each patient at two later time points—that is, after six and 12 months, respectively. Finally, at the end of sections II–IV, the treatment prescribed at each of these time points for the patient was to be specified.
Each participant was requested to follow scrupulously the guidelines furnished by us in completing the chart. These guidelines included a precise definition of each symptom and test according to the American Rheumatism Association's Dictionary of the Rheumatic Diseases.17 ,18 For certain items, more detailed definitions were provided by us (table3).
Definition of some of the items recorded in the clinical chart
DATA COLLECTION AND ANALYSIS
By the end of the enrolment period (March 1998) a total of 290 patients with SSc had been recruited, and participants had completed and returned sections I and II of the clinical charts for each of these patients. A statistical analysis of these preliminary data was carried out with the Statview program. Comparisons were made between patients attending the different centres based on analysis of variance, non-parametric tests for quantitative values, and contingency tables for proportions. In view of the high number of comparisons involved, we adopted a significance limit of 0.001.
Results
DEMOGRAPHIC AND HISTORICAL DATA
Table 4 summarises the main demographic features of the 290 patients recruited for this study. Women outnumbered men by 5.3 to 1, and there was a wide variation in the epidemiological parameters between patients. The disease duration, defined as the time elapsed from the onset of the first symptom (usually Raynaud's phenomenon), ranged from 0 to 56 years. When defined as the time elapsed from the first non-Raynaud manifestation, it ranged from 0 to 51 years. Thus a high percentage of the patients had disease of long duration. There was also in most cases a significant lapse in time between the patient's admission to the participating centre and his/her enrolment in the study; 190/290 had already been followed up at the respective centres for at least one year before being recruited into the study.
Main demographic features of the series of 290 patients with systemic sclerosis
As a result of the recruitment process, all the 290 patients fulfilled the ACR criteria for SSc (165 patients met the major criterion and 125 met two of the three minor criteria). One hundred and seventeen had the diffuse and 173 the limited form of the disease. Table 5 shows the demographic features of the patients by disease subset. A significant difference was seen in the mean disease duration, when evaluated from the onset of Raynaud's phenomenon (13 v 8 years), and in the mean age at enrolment (55v 49 years) between patients with lSSc and dSSc.
Main demographic features for the two disease subsets, limited and diffuse systemic sclerosis (SSc)
An analysis of variance showed a significant variation between centres in epidemiological parameters (fig 1), including the age at enrolment, the disease duration from the onset of the Raynaud's phenomenon, and the male to female ratio, which ranged from 1/2 to 1/20. In addition, significant differences emerged in subset distribution, the lSSc to dSSc ratio ranging from 5/1 to 1/2.5.

Distribution of the patients by (A) age at enrolment and (B) disease duration among the different centres. In the box plots the 10th, 25th, 50th (median), 75th, and 90th centiles for each variable are shown. Values above the 90th and below the 10th centiles are plotted as points.
Figure 2 shows the prevalence of organ involvement as reported in the patients' histories collected at the time of entry into the study. Significant differences (data not shown) emerged in the prevalence of various disease manifestations—namely, gut, lung, heart, kidney, joint, and muscle involvement, while the prevalence of skin sclerosis and Raynaud's phenomenon showed a slight difference only. Of course, a number of these differences must be ascribed to different investigative methods. Nevertheless, the detected differences in sex, age, subset distribution, and clinically detectable manifestations, such as joint involvement, point to actual diversities among different centre series.

Cumulative prevalence of organ involvement based on data from the patient history section—that is, collected at the time of entry into the study.
CLINICAL AND SEROLOGICAL FEATURES AT THE TIME OF ENROLMENT
Table 6 shows the clinical features reported for the 290 patients at enrolment. The most common findings were skin sclerosis, Raynaud's phenomenon, interstitial lung involvement, and oesophageal involvement. Skin sclerosis was found in 274 patients. Therefore, about 6% of the cases could be diagnosed as SSc without scleroderma. Kidney involvement (that is, either proteinuria or haematuria or increased creatininaemia) was detected in 52/290 (18%) of the patients, but no cases of renal crisis were recorded. Table 6 clearly shows that several items were characterised by a high number of missing values. This was particularly true for investigations to detect internal organ involvement. Despite these limitations, however, the figures for clinically detectable manifestations, such as scleredema, melanoderma, arthritis etc, must be considered reliable, each observer being an experienced clinician in scleroderma to whom clear cut guidelines had been provided.
Main clinical and serological features for the case series (at the time of the first observation)
When patients were compared by disease subset (dSScv lSSc), statistically significant differences in some of the clinical and serological manifestations were found (table 7; data shown in italics). Patients with dSSc had more severe skin involvement, associated with a higher frequency of functional impairment (that is, flexion contractures), tendon friction rubs, melanoderma, acro-osteolysis, digital necrosis, and reduced forced vital capacity and carbon monoxide transfer factor.
Prevalence of the different clinical and serological features among the two disease subsets
An analysis was carried out to elicit any differences between the centres in the prevalence of clinically detectable manifestations. A high degree of variability was found among centres in the reported frequency of various disease manifestations (figs 3 and 4). On the whole, the greatest variation was seen in the parameters of scleredema, melanoderma, calcinosis, and digital infarcts. As already stated, these items had been so carefully defined and actually are so easy to detect that the differences which emerged are to be considered true. On the other hand, the differences which emerged in the prevalences of interstitial lung involvement, oesophageal involvement, and autoantibody profile may depend on differing investigational tools.

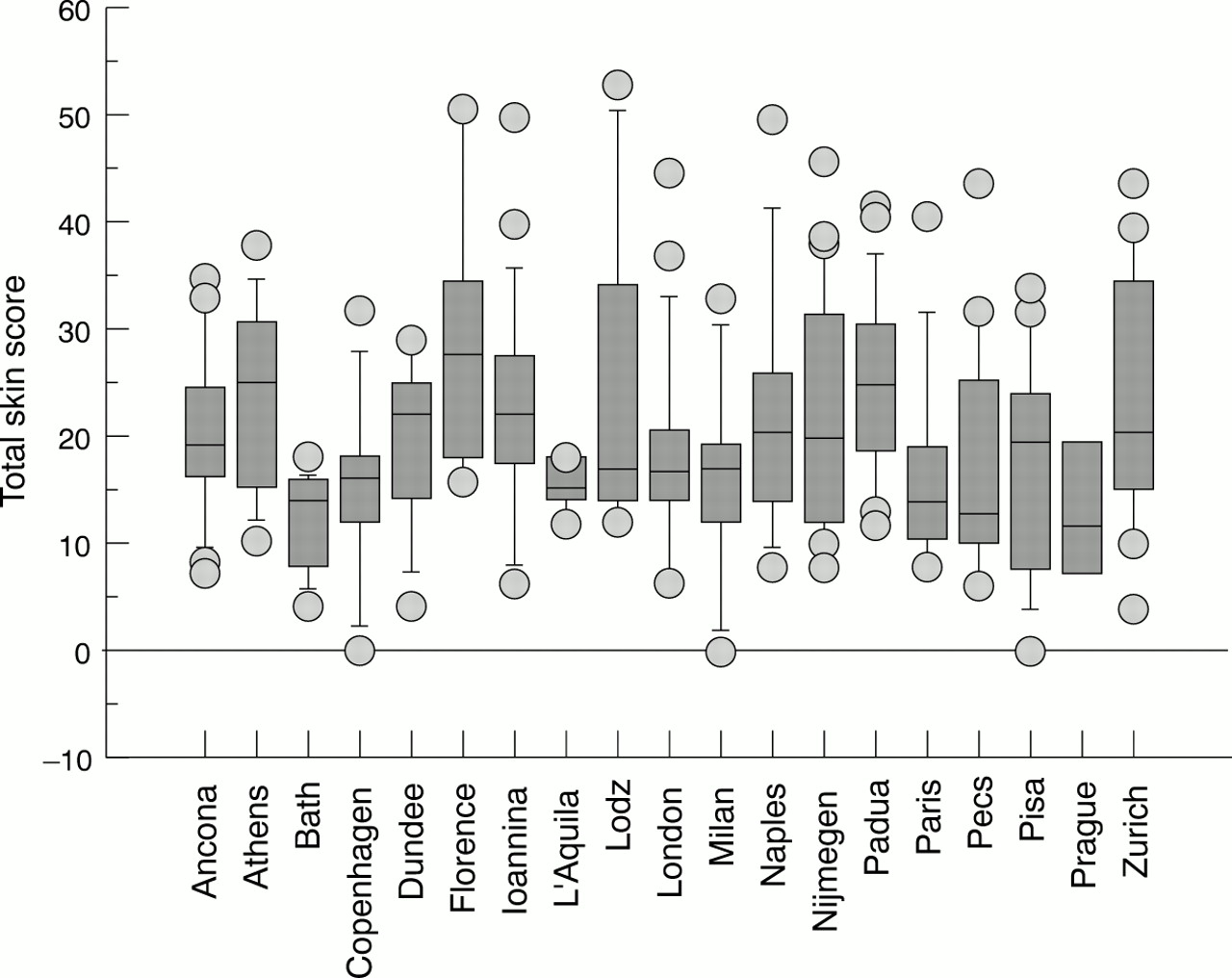
Distribution of the total skin score among the different centres. In the box plots the 10th, 25th, 50th (median), 75th, and 90th centiles for each variable are shown. Values above the 90th and below the 10th centiles are plotted as points.

Distribution of clinical features among the different centres as emerged by data collected in section II (clinical and instrumental investigations carried out at the entry into the study). In the box plots the 10th, 25th, 50th (median), 75th, 90th centiles for each variable are shown. Values above the 90th and below the 10th centiles are plotted as points.
TREATMENT
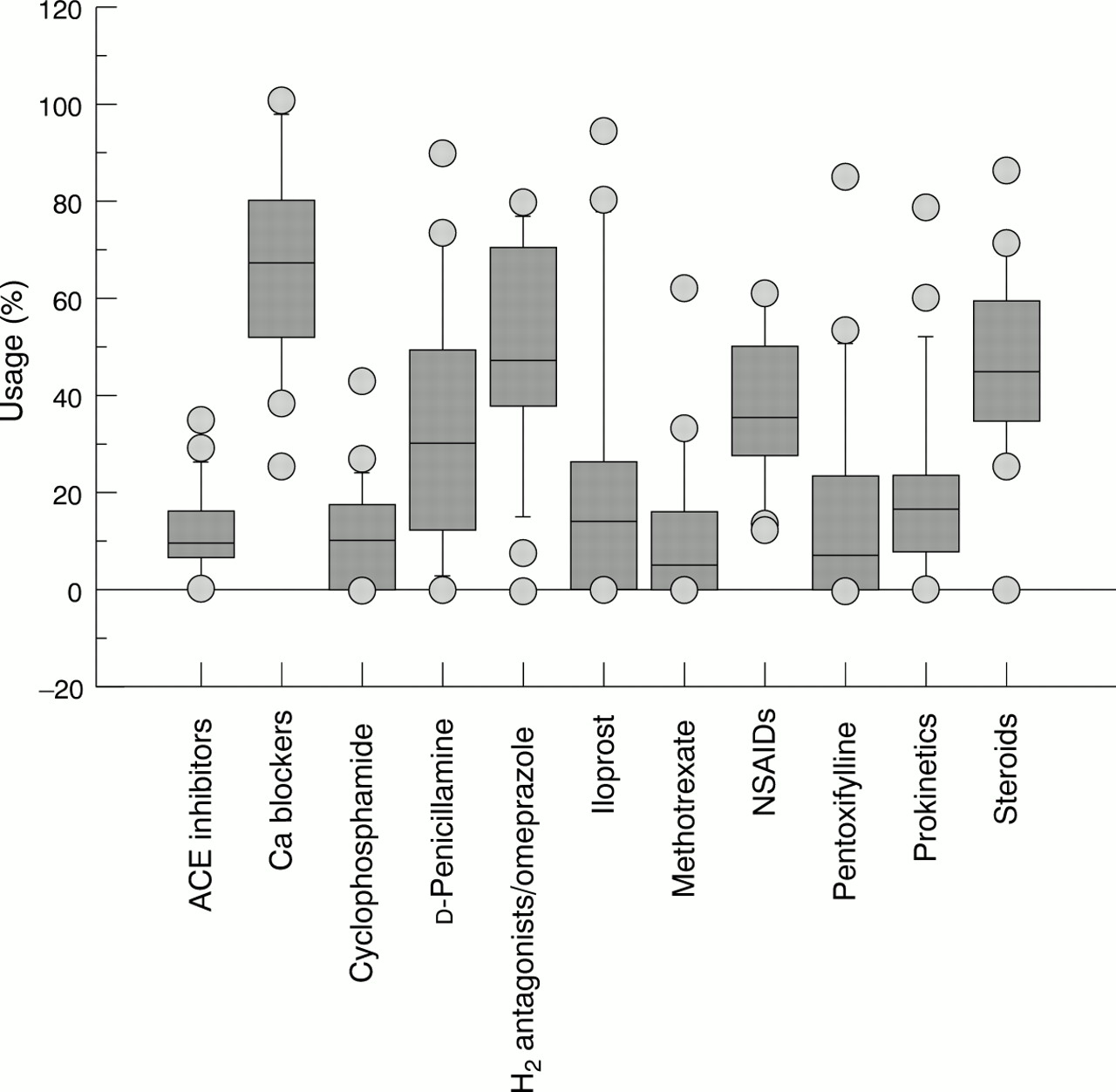
Figure 5 shows the treatment prescribed by the participating clinicians at the time of the first observation. The vast majority of the patients were being treated symptomatically with calcium channel blockers or other vasodilators, H2 antagonists, corticosteroids, or non-steroidal anti-inflammatory drugs. Only a small number of patients were receiving disease modifying drugs such as methotrexate, d-penicillamine, or cyclophosphamide. Considerable variation was seen in the therapeutic approach between different centres (fig 6). Only the use of H2antagonists/omeprazole was consistent across different centres. These results may depend on the differences referred to above among patients from various centres. Nevertheless, different therapeutic approaches also exist.

Drug treatment being used in the patient series at the time of enrolment.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Treatments used by the different centres. In the box plots the 10th, 25th, 50th (median), 75th, and 90th centiles of each variable are shown. Values above the 90th and below the 10th centiles are plotted as points.
Discussion
The aim of this multicentre European study, which is still continuing, will be to develop a valid set of disease criteria for SSc based on clinical data collected in a standardised manner on a large number of patients. Here we report the results of our analysis of the data gathered during the first phase of this study (patient history and evaluation at enrolment). The epidemiological and clinical serological findings for our series of 290 patients seem to be similar to data previously reported by others (tables 8 and 9). However, the data furnished by the 19 centres were far from homogeneous. The complex picture presented by SSc, characterised by a high variability in its clinical manifestations and treatment modalities, has already been discussed by other authors,12-15 ,27 though not in the light of rigorously designed, large scale studies.
Comparison of demographic, clinical, and serological data for the present series with similar data from other European series
Comparison of demographic, clinical, and serological data for the present series with similar data from other, non-European countries
Our prospective study, in which all of the centres were required to follow the same clearly defined protocol, clearly demonstrates the magnitude of the problem presented by this disease. The epidemiological variability observed as well as the high variability in the reported incidence of clinical features (such as calcinosis, scleredema, scleroderma, telangiectasia) may be ascribed to differences in the nature of the participating centres. It would be hard to believe that experienced clinicians might have failed to detect single manifestations such as melanoderma, digital necrosis, etc. On the other hand, the high number of missing values concerning most investigative methods prevents us from discussing the differences detected in the internal organ involvement. Nevertheless, the absence of patients with scleroderma renal crisis in our series deserves some comments. This complication has been identified as one of the leading causes of death in the North American series.8 ,28 ,29 However, studies conducted in Europe seem to indicate a lower prevalence.11 ,23 ,30 ,31 The absence of such a manifestation in our series may depend, however, on the nature of the study protocol, which enrolled consecutive patients and therefore might have excluded more severe cases. As far as kidney disease is concerned, we detected either proteinuria or haematuria or increased creatininaemia in 18% of our patients with SSc. However, we cannot confidently ascribe these alterations to SSc itself, because we did not correct the data for the presence of diabetes, hypertension, and other causes of these alterations.
In conclusion, certain observations may be made about SSc based on the preliminary results of our multicentre study. Despite our study design—which used a standardised chart that included all of the most generally accepted diagnostic parameters for SSc, carefully defined according to authoritative sources—we still found considerable differences between centres in the use of diagnostic tests and the therapeutic approach. This reflects the as yet unstandardised approach to SSc in different countries. To overcome these problems the scientific community will need to draw up a standardised procedure for the management of patients with SSc. This should include a precise definition of the major clinical parameters (including the establishment of a consensus on the more controversial aspects of the disease) and the determination of a series of simple and inexpensive diagnostic tests. This would provide a common research tool for different centres engaged in research on this complex disease.
European Scleroderma Study Group (EScSG)
G Valentini, S D'Angelo, A De Luca, E Tirri (Second University of Naples, Italy); S Bombardieri (chairman), A Della Rossa, W Bencivelli, C Ferri (Clinical Immunology/Rheumatology Units, Department of Internal Medicine, University of Pisa, Italy); M Matucci Cerinic, S Generini (Istituto di Clinica Medica IV, University of Florence, Italy); AJ Silman (University of Manchester, UK); JF Belch (Ninewells Hospital and Medical School, Dundee, UK); CM Black (Royal Free Academy Hospital, London, UK); P Bruhlman, S Enderlin (University Hospital, Zurich, Switzerland); L Czirják (University of Pecs, Hungary); AA Drosos (University of Ioannina, Greece); G Danieli, A Gabrielli, S Tartari De Carolis (University of Ancona, Italy); G Tonietti, R Giacomelli, P Cipriani (University of L'Aquila, Italy); M Inanc (University of Istanbul, Turkey); NJ McHugh (Royal National Hospital for Rheumatic Disease, Bath, UK); O Meyer, G Hayem (University of Paris VII, Paris, France); H Nielsen (Rheumatology Unit, Herlev Hospital, Herlev, Denmark); R Scorza, S Bazzi, M Carroni (University of Milan, Italy); J Stork, R Becvar (Charles University, Prague, Czech Republic); A Sysa, B Dziankowska (University of Lodz, Poland); S Todesco, M Rosada, F Cozzi (University of Padua, Italy); FHJ van den Hoogen (University Hospital, Nijmegen, The Netherlands); PG Vlachoyannopoulos (National University, Athens, Greece).
References
Footnotes
↵* Members of the European Scleroderma Study Group are given in the .