Article Text

Abstract
OBJECTIVE To examine the hypothesis that fetal microchimerism plays a part in the pathogenic process of Sjögren's syndrome (SS).
METHODS Genomic DNA samples were extracted from peripheral blood whole nucleated cells and the CD34+ cell enriched fraction of patients with SS and healthy women who had male offspring as well as nulliparous women. A Y chromosome-specific sequence was detected as a marker for fetal cells by a nested polymerase chain reaction (PCR) and by DNA hybridisation combined with PCR using specific primers and probes. All procedures were performed with great care to avoid the contamination of male DNA.
RESULTS A nested PCR and DNA hybridisation combined with PCR was established that can detect a single male cell out of 1.67×105 female cells. It was not possible to increase the sensitivity further because the amount of template DNA held in the PCR was limited. When these methods were used, no fetal cells were detected in any samples from patients with SS, though they were detected in whole nucleated cells from two healthy women who had delivered sons previously.
CONCLUSIONS The findings indicate that circulating fetal cells in patients with SS are uncommon ( < 1 in 1.67×105), if they exist. With the conventional PCR based methods that were used, it is difficult to evaluate the quantitative difference in circulating fetal cells between patients with SS and healthy women.
- microchimerism
- graft versus host disease
- scleroderma
- Sjögren's syndrome
Statistics from Altmetric.com
Sjögren's syndrome (SS) is an autoimmune disease characterised by lymphocytic infiltration into lachrymal and salivary glands, leading to a significant decrease in tear and saliva secretion.1 This disorder is associated with the production of various autoantibodies.2 ,3 Although previous studies have shown that the pathogenic process of SS may be associated with numerous factors of viral, endocrine, neural, genetic, and environmental origin,4-7 the mechanism responsible for development of the disease still remains unclear.
Another consideration in understanding the cause of SS is the markedly higher incidence in women—more than nine times as common in women than in men.8 A number of studies have described the effect of sex hormones on the female predisposition of autoimmune diseases, including SS.5 ,9-12 Androgens dramatically suppress lymphocytic infiltration in lachrymal and salivary glands of female MRL/lpr mice.11 In contrast, prenatal exposure to oestrogens in normal mice induced salivary gland lesions indistinguishable from SS and accelerated autoantibody production.10 However, the effect of these hormone actions is controversial.13 Thus sex hormones may be a risk factor for the development of SS, but hormonal influence alone is not sufficient to explain the female predominance in SS.
Another possible factor associated with the female predominance in SS is pregnancy. It is now evident that pregnancy is an important immunological event in the life of women. It has been recognised that non-self fetal cells migrate into the maternal circulation during pregnancy.14 Among these cells, fetal haematopoietic progenitor cells have been reported to persist in the maternal circulation for decades.15 Thus these mothers are in a condition of microchimerism, in which two genetically different cells co-exist in one person.16 Symptoms of SS are similar to those of chronic graft versus host disease (GVHD) caused by the chimeric condition after allogeneic stem cell transplantation (SCT).17 ,18 The characteristic hallmark shared by SS and chronic GVHD is progressive atrophy and dryness of the mucosal surface.19 ,20 A lachrymal or salivary gland biopsy sample from patients with chronic GVHD showed lymphocytic infiltration, similar to that found in SS.
Other autoimmune diseases that predominantly affect women after childbearing age and have clinical similarities to chronic GVHD include scleroderma and primary biliary cirrhosis.19 ,21 Recently, Nelson proposed the hypothesis that fetal microchimerism plays a part in the pathogenesis of scleroderma.22 In her theory, some cases of scleroderma in women with children are actually GVHD caused by fetal cells. This hypothesis was supported by two independent reports that described an increased incidence of circulating fetal cells and also detected fetal cells in skin lesions from female patients with scleroderma.23 ,24 In this report, to examine the possible role of microchimerism in the development of SS, we have attempted to detect circulating fetal cells in patients with SS using carefully established polymerase chain reaction (PCR) based assays.
Patients and methods
PATIENTS AND CONTROLS
Thirty female patients with SS who had delivered at least one son were enrolled in this study. All patients satisfied the European criteria for SS25 and had no additional collagen disease (primary SS). Eighteen healthy women who had sons and eight nulliparous women served as controls. These healthy women had no self reported autoimmune disease or history of previous blood transfusion. The mean age of the subjects at blood collection was 60 (range 36–68) in patients with SS, 47 (34–56) in healthy women with sons, and 25 (22–36) in nulliparous women. The mean period between delivery of a male child and blood sampling was 29.3 years (3–50) in patients with SS and 18.1 years (7–24) in healthy women with sons (p=0.01). One male healthy donor and a female SCT recipient from a male donor were used as positive controls in assays for the detection of male cells. Three women who had delivered male children within the past 24 hours were used to validate the sensitivity of our detection methods. Informed consent was obtained from all subjects before blood collection.
PREPARATION OF GENOMIC DNA
Peripheral blood whole nucleated cells were separated from 20 ml heparinised venous blood in the presence of 0.6% dextran. CD34+ cells were enriched from peripheral blood mononuclear cells (1–4×107) by incubating with 4×107magnetic beads coated with monoclonal antibody against human CD34 (Dynal, Oslo, Norway). In some samples, the collected cells were detached from the beads, stained with the FITC conjugated anti-CD34 antibody, and analysed on a FACScalibur flow cytometer (Becton Dickinson, Bedford, MA) to determine the efficiency of the technique. Genomic DNA was extracted from peripheral blood whole nucleated cells or a CD34+ cell enriched fraction by digestion with proteinase K, followed by phenol extraction and ethanol precipitation.
NESTED PCR FOR DETECTION OF MALE CELLS
Male DNA was detected by amplifying genomic DNA by nested PCR with primers specific for a Y chromosome-specific gene, TSPY (testis-specific protein Y encoded).26 TSPY is located on Yp and has no known homology with autosomal genes. The first PCR was conducted using a sense primer (5′-CTTCCACC TTCAGCCACCGCCTCCTCT-3′) and an antisense primer (5′-CTGTTGTGCGC TGCCTTGACGACCCAG-3′). The first PCR comprised 40 cycles of 30 seconds at 95°C, 30 seconds at 66°C, and one minute at 72°C. The reaction mixture contained template DNA (1 μg, except as indicated otherwise), 2 mM MgCl2, 200 μmol/l each of dNTPs, 2.5 units of Ampli-Taq Gold (Perkin-Elmer Cetus, Foster City, CA), and 1 μmol/l of each of the sense and antisense primers in 50 μl of total volume. The second amplification of 20 cycles was performed using 2 μl of the first PCR product with a sense primer (5′-AGCATTGCCTCCAGCCTGAA-3′) and an antisense primer (5′-AGCGGTTGCGGT GCCTGTTG-3′). The PCR amplified products were subjected to electrophoresis on a 1.5% agarose gel, followed by ethidium bromide staining. All PCR experiments contained a negative control (without template DNA) and positive controls (male DNA and DNA from a female SCT recipient from a male donor). All procedures were performed with great care to avoid contamination by male DNA. For example, experiments were performed in an environment isolated from men and all reagents were freshly prepared in every experiment.
In some experiments the PCR products were directly sequenced on the ABI Prism 310 Genetic Analyzer (Applied Biosystems, Foster City, CA) using the BigDye Terminator Cycle Sequence Ready Reaction kit.
DNA HYBRIDISATION COMBINED WITH PCR FOR DETECTION OF MALE CELLS
The products of the first PCR were also applied to DNA hybridisation using digoxigenin labelled oligonucleotide probes specific for the TSPY sequence. The PCR products fractionated on an agarose gel were transferred onto a nylon membrane (Hybond-N+; Amasham Pharmacia Biotech, Piscataway, NJ) by capillary blotting and hybridised with the probes at 52°C overnight. A mixture of five probes corresponding to different portions of the TSPY gene (5′-GCCAGCCCAGC CCAG-3′, 5′-CCCCAGCAGACCCGC-3′, 5′-CCCGCAGATCCCGCA-3′, 5′-AGGCACC GCAACCGC-3′, and 5′-CTGCGGGTG CGGAG-3′) was used to increase the sensitivity. The membrane was washed and then processed using a commercially available detection system (DIG DNA detection kit; Boehringer Mannheim, Indianapolis, IN).
Results
ESTABLISHMENT OF NESTED PCR AND DNA HYBRIDISATION COMBINED WITH PCR FOR DETECTION OF MALE CELLS
Because the number of male cells in the female circulation was expected to be low, it was necessary to increase the amount of template DNA in PCR as much as possible. To determine the amount of template DNA that gives the highest number of copies of the product of interest without changing the PCR efficiency, the first PCR was carried out using a mixture of serial amounts of background female genomic DNA (100 ng—10 μg) and 10 ng male DNA as templates (fig 1). The amplification occurred at the same efficiency in the presence of 100 ng, 300 ng, 600 ng, and 1 μg of background female DNA. When ⩾2 μg of template DNA was added to the tubes for PCR, the PCR amplification efficiency decreased. In view of this result, 1 μg of genomic DNA was used as a template in the following experiments.

Amplification of the TSPY gene using male DNA as a template in the presence of serial amounts of background female DNA. Serial amounts of female genomic DNA (100 ng—10 μg) were mixed with 10 ng male genomic DNA and used as templates for the first polymerase chain reaction (PCR). The PCR products were fractionated on a 1.5% agarose gel and stained with ethidium bromide. Lane 1: 100 ng, lane 2: 300 ng, lane 3: 600 ng, lane 4: 1 μg, lane 5: 2 μg, lane 6: 5 μg, and lane 7: 10 μg of background female DNA. Lane 8: molecular weight markers (100 bp ladder).
To assess the sensitivity of detection of male DNA, serial amounts of male DNA (10−3–10−10 μg) mixed with 1 μg of female DNA were amplified in a nested PCR (fig 2). A Y chromosome-specific 345 bp fragment was detectable when ⩾10−7 μg of male DNA was used as a template. PCR using 10−6 μg and 10−7 μg of male DNA gave negative results in repeat experiments, whereas PCR using ⩾10−5 μg of male DNA constantly showed positive results. DNA sequencing confirmed that the 345 bp fragment had a nucleotide sequence identical to that of the TSPY gene. To evaluate further the reliability of our detection system, a nested PCR was conducted using genomic DNA obtained from three women who had delivered sons within the past 24 hours. As shown in fig 3, a Y chromosome-specific product was detected in one of three samples.

Detection of the TSPY gene by nested polymerase chain reaction (PCR) using serial amounts of male DNA as templates. Serial amounts of male DNA (10−3–10−10 μg) in 1 μg of female DNA were amplified by the nested PCR. The PCR products were fractionated on 1.5% agarose gel and stained with ethidium bromide. Lane 1: 10−3 μg, lane 2: 10−4μg, lane 3: 10−5 μg, lane 4: 10−6 μg, lane 5: 10−7 μg, lane 6: 10−8 μg, lane 7: 10−9 μg, and lane 8: 10−10 μg of male DNA. Lane 9: no template for the first PCR and lane 10: no template for the second PCR. Lane 11: molecular weight markers (100 bp ladder).
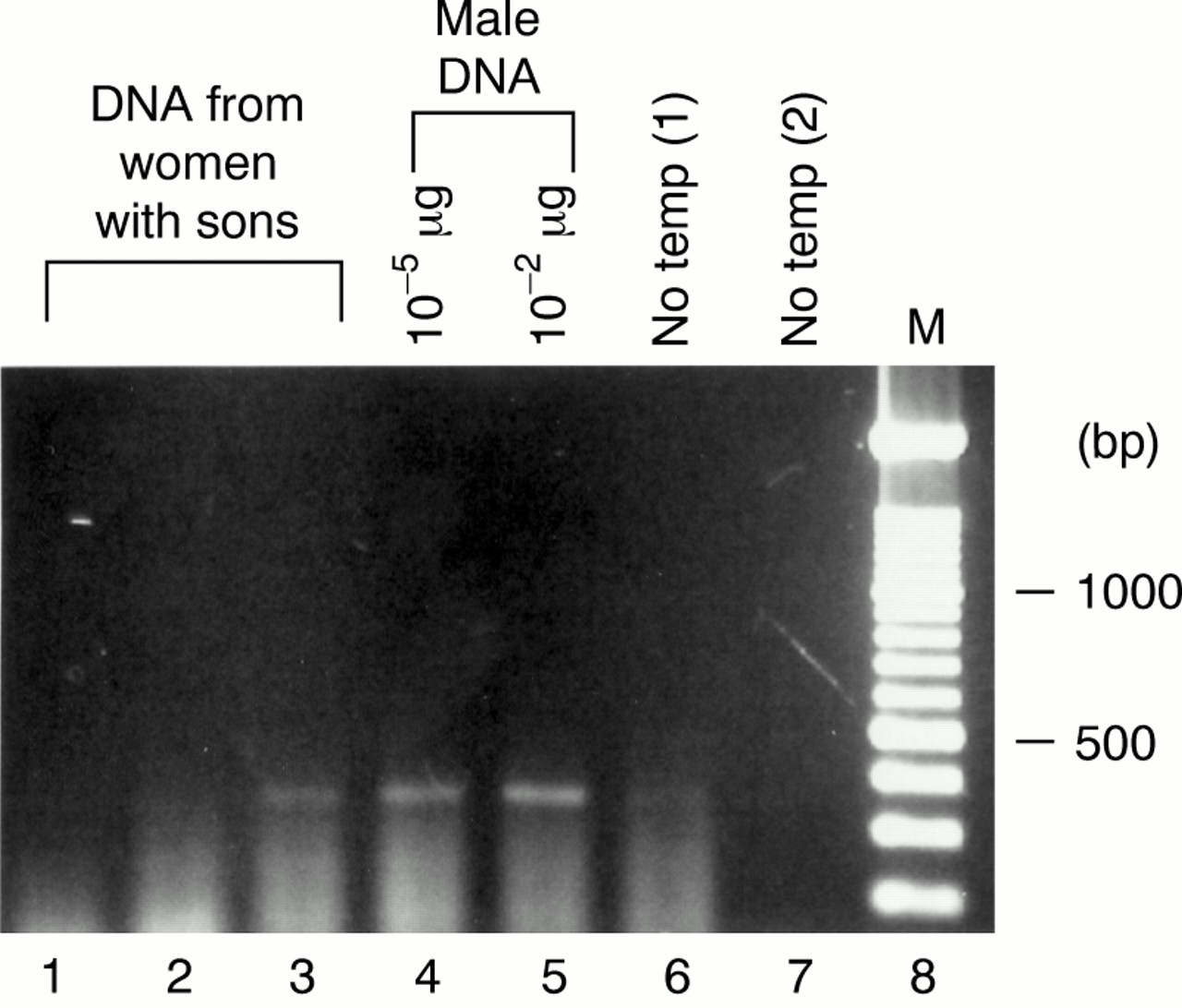
Detection of male cells in peripheral blood whole nucleated cells from women who had delivered sons within the past 24 hours by a nested polymerase chain reaction (PCR). Genomic DNA from peripheral blood whole nucleated cells obtained from three women one day after delivery (lanes 1–3: 1 μg), and control male DNA (lane 4: 10−5 μg and lane 5: 10−2 μg) were applied for the nested PCR. The PCR products were fractionated on 1.5% agarose gel and stained with ethidium bromide. Lane 6: no template for the first PCR and lane 7: no template for the second PCR. Lane 8: molecular weight markers (100 bp ladder).
The PCR products of the first PCR using serial amounts of male DNA were then hybridised with the TSPY probes. Positive signals were always detected when ⩾10−5 μg of male DNA was used in the first PCR (data not shown), indicating that the sensitivity for the detection of male DNA was similar for the nested PCR and the DNA hybridisation combined with PCR.
DETECTION OF FETAL CELLS IN CIRCULATING WHOLE NUCLEATED CELLS
Genomic DNA samples from peripheral blood whole nucleated cells of 18 patients with SS with sons, 12 healthy women with sons, and five healthy women with no sons were analysed by nested PCR (fig 4). Male cells in the circulation of previously pregnant women were considered as fetal cells. The experiment was repeated three times, but no fetal cells were detected in any samples.
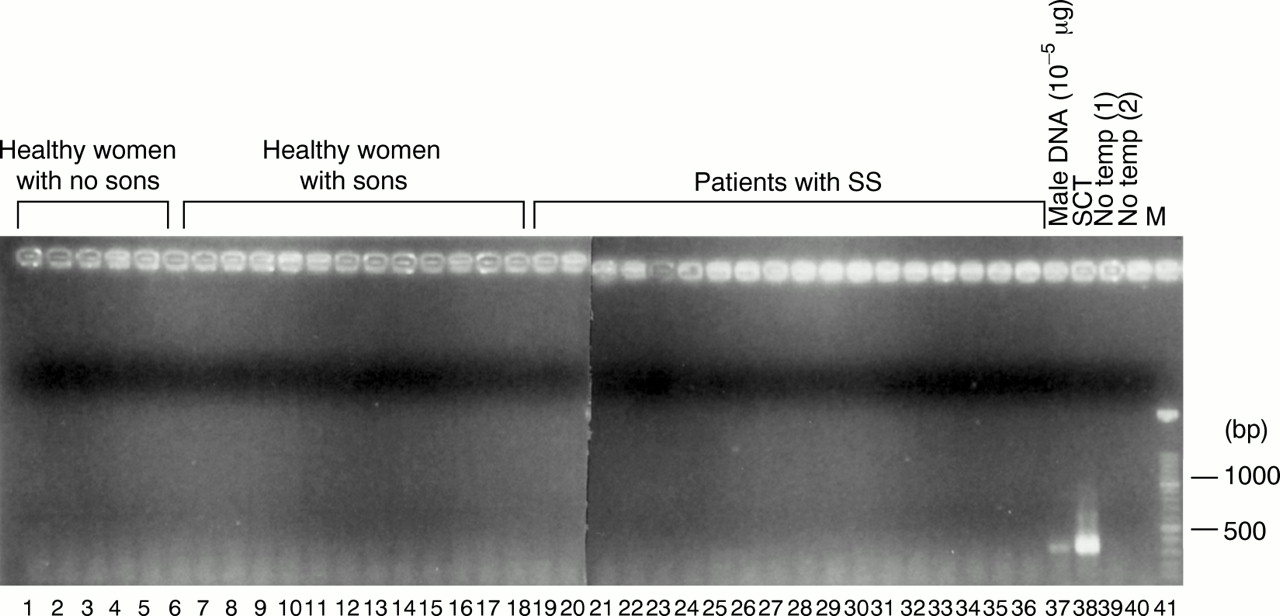
Detection of male cells in peripheral blood whole nucleated cells from healthy women with no sons, healthy women with sons, and patients with SS with sons by a nested polymerase chain reaction (PCR). Genomic DNA (1 μg) from these subjects was amplified by a nested PCR, and the PCR products were fractionated on 1.5% agarose gel and stained with ethidium bromide. Lanes 1–6: healthy women with no sons, lanes 7–17: healthy women with sons, and lanes 18–36: patients with SS with sons. Lane 37: male DNA (10−5 μg), lane 38: DNA from a female stem cell transplantation (SCT) recipient from a male donor (1 μg), lane 39: no template for the first PCR, and lane 40: no template for the second PCR. Lane 41: molecular weight markers (100 bp ladder).
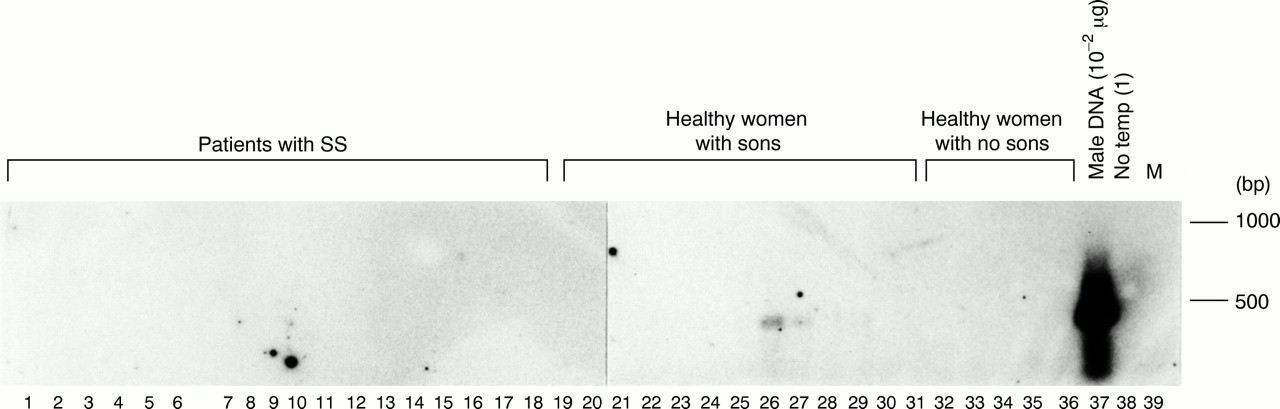
A DNA hybridisation combined with PCR was also carried out using the same DNA samples on three different occasions (fig 5). Fetal cells were detected in two healthy women with sons in one experiment, but these samples gave negative results in the other two experiments. No positive signal was detected in any samples from patients with SS in the three experiments.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Detection of male cells in peripheral blood whole nucleated cells from patients with Sjögren's syndrome with sons, healthy women with sons, and healthy women with no sons by DNA hybridisation combined with a polymerase chain reaction (PCR). The PCR products of the first PCR were fractionated by electrophoresis and hybridised with probes specific for the TSPY gene. Reactivities were visualised with a chemiluminescence detection system. Lanes 1–18: patients with SS with sons, lanes 19–31: healthy women with sons, and lanes 32–36: healthy women with no sons. Lane 37: male DNA (10−2 μg) in the first PCR and lane 38: no template for the first PCR. Lane 39: molecular weight markers (100 bp ladder).
DETECTION OF FETAL CELLS IN CIRCULATING CD34+ CELL ENRICHED FRACTION
The presence of fetal cells in the CD34+ cell enriched fraction from 10 patients with SS with sons, five healthy women with sons, and three healthy women with no sons was examined by a nested PCR and by DNA hybridisation combined with PCR. Flow cytometric analysis showed that the CD34+ cell enriched fraction obtained from two samples contained 10% and 12% CD34+ cells, indicating that CD34+ cells were enriched by at least 10 times. Fetal cells were detected in none of the samples either by the nested PCR or by the DNA hybridisation combined with PCR (data not shown).
Discussion
In this study we failed to obtain evidence to show that fetal microchimerism is increased in peripheral blood whole nucleated cells or the CD34+ cell enriched fraction of patients with SS using carefully established PCR based methods for the detection of male cells. Circulating fetal cells were detected in two healthy women with sons, but not in any patients with SS. Therefore, the incidence of fetal cells in the circulation was too low to assess a quantitative difference between patients with SS and healthy subjects by our methods.
It is unlikely that the failure to detect fetal cells in this study is due to the low sensitivity of our assays. We used 1 μg of genomic DNA for the first PCR, because this was the maximum quantity that can be used in PCR as a template without inhibition of the amplification efficiency. One microgram of genomic DNA is equivalent to the amount of DNA that is contained in 1.67×105 cells, based on an assumption that a single cell contains approximately 6 pg of genomic DNA.27 Male DNA was constantly detected in the nested PCR using ⩾10−5 μg male DNA mixed with 1 μg of background female DNA as templates. In contrast, a Y chromosome-specific product was sometimes detected when <10−6 μg of male cells was added in the PCR, probably because 1 μg of genomic DNA contained male DNA by chance in these dilutions. This finding indicated that sensitivity of our nested PCR exceeded one male cell out of 1.67×105 female cells and, therefore, our nested PCR can detect a single male cell in 1.67 × 105 female cells. This sensitivity was confirmed by the finding that male DNA was detected in one of three women who had delivered sons within the past 24 hours; a previous report showed that fetal cells were often detected when 105 nucleated cells from maternal peripheral blood were assessed one day after delivery.14 The sensitivity of the nested PCR was similar to the DNA hybridisation combined with PCR, and was comparable with that in a previously reported method for the detection of the Y chromosome-specific sequence.28 Therefore, our results indicate that circulating fetal cells, if they exist, are fewer than 1 out of 1.67×105 host cells in patients with SS and in healthy women who were previously pregnant.
Two recent reports showed that the incidence of fetal cells in peripheral blood from patients with scleroderma was higher than in blood from control women.23 ,24 The discrepancy between our finding and these results may merely reflect the difference in the pathogenic process between scleroderma and SS, but it may be due to several problems in the methods they used for the detection of fetal cells. In fact, one recent report failed to confirm their results using the same detection method.29 These investigators used PCR based methods to detect the Y chromosome-specific sequence as a marker for fetal cells. The incidence of circulating fetal cells in their reports appeared to be less than one out of 106 cells, though they did not mention the amount of genomic DNA used in the PCR as a template. Another problem is that the Y chromosome sequence was detected even in women who had no sons.30 Possibly, male cells or DNA contaminated the experiments. Nested PCR and PCR using radiolabelled nucleotides are so sensitive that they can detect even one copy of contaminating male DNA. Therefore, any contact with men and their belongings should be avoided in the experiments. We ourselves have sometimes obtained false positive results in negative control samples after repeated use of reagents, possibly due to contamination by male DNA, despite great care to prevent contamination.
To increase the sensitivity for detection of circulating fetal cells, a CD34+ cell enriched fraction was used in our assays, based on a previous finding that fetal CD34+ cells preferentially persist in maternal peripheral blood for decades after pregnancy.15However, fetal cells were below the detectable level even when the CD34+ cell enriched fraction was used, though the number of samples examined was small. Recently, Evans et alshowed that fetal DNA was detected in various subsets of peripheral blood mononuclear cells, such as T cells, B cells, NK cells, and monocytes, from some patients with scleroderma and healthy women.30 Taken together, our findings indicate that fetal cells are not concentrated in the CD34+ cell population, but, rather, are distributed in various subsets of maternal circulation after pregnancy.
The incidence of circulating non-self cells is expected to be low in patients with SS, if they exist (<1 in 1.67×105), whereas cells derived from bone marrow are totally exchanged in patients with chronic GVHD after allogeneic SCT. Recently, we have found that ultrastructural findings in the lachrymal gland are significantly different between SS and chronic GVHD.31Thus because of a remarkable difference in the number of effector cells and histological findings of these two diseases, the pathogenic process of SS and chronic GVHD may not be mediated by the same mechanism.
Is then the microchimerism theory for the development of SS totally irrelevant? The incidence of fetal cells in maternal peripheral blood is low whether the subjects have SS or not, but the possibility cannot be excluded that a significant difference might be found if more sensitive assays than the conventional PCR based methods were available and used. Because the incidence of fetal cells is influenced by the period since the last delivery,14 patients with SS might have had a higher incidence of fetal cells immediately after delivery. Moreover, it is still possible that a small number of fetal cells play a part in the pathogenesis of SS. Fetal cells may migrate preferentially into target organs of the disease rather than into the circulation. Artlett et al used fluorescence in situ hybridisation to identify a Y chromosome sequence in skin biopsy specimens from women with scleroderma who had male children.24 To date, fetal cells in lachrymal or salivary glands have not been examined in patients with SS, but a recent study analysing 10 liver specimens from women with primary biliary cirrhosis failed to detect the Y chromosome sequences in any samples.32 Another possibility is that antigen-presenting cells and immune regulatory cells derived from fetal haematopoietic stem cells disregulate the maternal immune system, resulting in induction and/or promotion of autoimmunity.
Acknowledgments
We thank Drs Nobuaki Ozawa and Yasunori Yoshimura for providing peripheral blood samples from women who gave birth to male children one day previously, and Ms Yuka Okazaki for assisting in the flow cytometric analysis.
References
Footnotes
This work was supported by the grants from the Japanese Ministry of Health and Welfare and from the Japanese Ministry of Education, Science, and Culture.