Article Text

Abstract
Objective: The phase III GO-FORWARD study examined the efficacy and safety of golimumab in patients with active rheumatoid arthritis (RA) despite methotrexate therapy.
Methods: Patients were randomly assigned in a 3 : 3 : 2 : 2 ratio to receive placebo injections plus methotrexate capsules (group 1, n = 133), golimumab 100 mg injections plus placebo capsules (group 2, n = 133), golimumab 50 mg injections plus methotrexate capsules (group 3, n = 89), or golimumab 100 mg injections plus methotrexate capsules (group 4, n = 89). Injections were administered subcutaneously every 4 weeks. The co-primary endpoints were the proportion of patients with 20% or greater improvement in the American College of Rheumatology criteria (ACR20) at week 14 and the change from baseline in the health assessment questionnaire-disability index (HAQ-DI) score at week 24.
Results: The proportion of patients who achieved an ACR20 response at week 14 was 33.1% in the placebo plus methotrexate group, 44.4% (p = 0.059) in the golimumab 100 mg plus placebo group, 55.1% (p = 0.001) in the golimumab 50 mg plus methotrexate group and 56.2% (p<0.001) in the golimumab 100 mg plus methotrexate group. At week 24, median improvements from baseline in HAQ-DI scores were 0.13, 0.13 (p = 0.240), 0.38 (p<0.001) and 0.50 (p<0.001), respectively. During the placebo-controlled portion of the study (to week 16), serious adverse events occurred in 2.3%, 3.8%, 5.6% and 9.0% of patients and serious infections occurred in 0.8%, 0.8%, 2.2% and 5.6%, respectively.
Conclusion: The addition of golimumab to methotrexate in patients with active RA despite methotrexate therapy significantly reduced the signs and symptoms of RA and improved physical function.
Statistics from Altmetric.com
Clinical studies have demonstrated that treatment with biological agents that target tumour necrosis factor (TNF) improve manifestations of rheumatoid arthritis (RA).1–10 These compounds have been shown to provide greater benefit when combined with methotrexate therapy than when each agent or methotrexate is administered alone.10–12 However, all currently available anti-TNFα agents have differences in affinity, stability, solubility, terminal half-life characteristics and dosing regimens.13–15
Golimumab is a human anti-TNFα monoclonal antibody that was generated and affinity matured in an in-vivo system. Golimumab has a high affinity and specificity for human TNFα and effectively neutralises TNFα bioactivity in vitro.16 Results of an earlier phase II study of golimumab in 172 patients with active RA despite methotrexate therapy demonstrated the efficacy of golimumab given every 4 weeks by subcutaneous injection in combination with methotrexate.17 The clinical effect was evident within 2 weeks of the first dose and was sustained to 1 year. In this larger phase III study, we evaluated the efficacy and safety of golimumab in the treatment of patients with active RA despite methotrexate therapy.
PATIENTS AND METHODS
This was a phase III, multicentre, randomised, double-blind, placebo controlled trial. The study included a double-blind controlled phase to week 52 and an open-label extension up to 5 years. In this report, we present the results to week 24, which include the co-primary endpoints at weeks 14 and 24. Patients were enrolled at 60 investigational sites in 12 countries: Argentina, Australia, Canada, Chile, Germany, Hungary, Mexico, New Zealand, Poland, South Korea, Taiwan and the USA.
The study was conducted in accordance with the Declaration of Helsinki and good clinical practices. The protocol was reviewed and approved by each site’s institutional review board or ethics committee. All patients provided written informed consent before undergoing study-related procedures.
Patients
Study participants were 18 years of age or older, had a diagnosis of RA according to the revised 1987 criteria of the American College of Rheumatology (ACR)18 for at least 3 months before screening, and were to have been on a stable methotrexate dose of 15 mg/week or greater but 25 mg/week or less during the 4-week period immediately preceding screening. Patients were to have tolerated 15 mg/week or greater of methotrexate for at least 3 months before screening. Patients were required to have active RA, defined as four of more swollen joints (out of 66 total) and four or more tender joints (out of 68 total) and at least two of the following: (1) C-reactive protein (CRP) of 1.5 mg/dl or greater (normal range 0–0.6 mg/dl) or erythrocyte sedimentation rate (ESR) by the Westergren method of 28 mm/h or greater; (2) at least 30 minutes of morning stiffness; (3) bone erosion determined by x ray and/or magnetic resonance imaging; or (4) anti-cyclic citrullinated peptide antibody or rheumatoid factor positive test results. Eligible patients had to have met the tuberculosis screening criteria (supplemental material 1, available online only).
Patients who were using non-steroidal anti-inflammatory drugs or other analgesics for RA had to be taking a stable dose for at least 2 weeks before the first dose of study agent. Patients who were taking oral corticosteroids had to have been receiving a stable dose equivalent to 10 mg/day or less of prednisone for at least 2 weeks before the first dose of study agent.
Patients were excluded from study participation if they had a known hypersensitivity to human immunoglobulin proteins or other components of golimumab. Any previous use of any anti-TNF agent, rituximab, natalizumab or cytotoxic agents excluded patients from study participation. In addition, patients should not have received anakinra; disease-modifying antirheumatic drugs other than methotrexate; or intravenous, intramuscular, or intra-articular corticosteroids within 4 weeks before the first dose of study agent or alefacept or efalizumab within 3 months before the first dose of the study agent. A complete list of inclusion and exclusion criteria is provided in supplemental material 1 (available online only).
Study design
During a 4-week run-in period between screening and randomisation, all patients received study-supplied over-encapsulated methotrexate capsules at their pre-study stable dose. Patients were re-evaluated for study eligibility at the end of this 4-week run-in period. Patients who still met the study criteria were randomly assigned in a 3 : 3 : 2 : 2 ratio to receive: placebo injections plus methotrexate (group 1), golimumab 100 mg injections plus placebo capsules (group 2), golimumab 50 mg injections plus methotrexate (group 3), or golimumab 100 mg injections plus methotrexate (group 4).
Randomisation was stratified by investigational site and was conducted using a telephone interactive voice response system. All patients continued their initial stable doses of methotrexate throughout the study without interruption except for patients in group 2 who were switched from active methotrexate to sham methotrexate capsules at randomisation without any washout period before study entry.
At week 16, patients in groups 1, 2 or 3 with less than a 20% improvement from baseline in both tender and swollen joint counts had their study medication adjusted in a double-blind fashion (ie, early escape). Patients in group 1 who met the criteria for early escape began receiving active golimumab 50 mg every 4 weeks and continued their stable dose of active methotrexate. Patients in group 2 who met the criteria for early escape had their sham methotrexate capsules replaced with active methotrexate capsules at the same stable dose that the patient had received at screening and continued to receive injections of golimumab 100 mg every 4 weeks. Patients in group 3 who met the criteria for early escape had their golimumab dose increased from 50 mg to 100 mg every 4 weeks and continued to receive their stable dose of active methotrexate. Patients in group 4 had no adjustments made to their subcutaneous or oral study medication.
Treatments
Centocor Research and Development, Inc supplied all study medication. Golimumab and placebo were supplied as sterile liquid for subcutaneous injection. Placebo injections contained the same solution as active golimumab but did not contain the monoclonal antibody. Active methotrexate and placebo methotrexate were supplied as identical opaque capsules. Injections were administered every 4 weeks and each patient received two injections per dose (0.5 ml and 1.0 ml syringes) to maintain the blind.
Evaluations
Response to treatment was assessed using the ACR response criteria (ACR20/50/70).19 ACR-N9 20 was also calculated. The disease activity score in 28 joints (DAS28)21 was calculated separately using both CRP and ESR. The European League Against Rheumatism (EULAR) response was calculated according to previously described methods,22 and DAS28 remission was defined as a score of less than 2.6.22
An independent assessor designated at each study centre performed all joint assessments. The joint assessor had no other contact with the patient, was not the treating physician and was not permitted to review patient medical records, case report forms, or previous joint counts during the study.
The health assessment questionnaire disability index (HAQ-DI) was used to evaluate physical function.23 The minimum clinically important difference in HAQ-DI score has previously been defined as a reduction of 0.22 or greater.24 We evaluated the proportions of patients with a reduction from baseline in HAQ-DI of 0.25 or greater as a more conservative estimation of the minimum clinically important change.
Serum samples taken at baseline and week 24 were assessed for the presence of antibodies to golimumab using a previously described assay.17 The presence of the study agent in the serum interferes with the detection of antibodies to the study agent in these types of assays.11 25
Sample size calculation
Assuming 55% or more of patients in groups 3 and 4 and 35% of patients in group 1 would achieve an ACR20 response, a sample size of 120 patients in group 1 and 80 patients in groups 3 and 4 was required to achieve greater than 90% power (two-sided χ2, α = 0.05). Assuming 55% of patients in group 2 and 35% of patients in group 1 would achieve an ACR20 response, a sample size of 120 patients in both groups 1 and 2 was needed to achieve greater than 85% power (two-sided χ2 test, α = 0.05). This sample size would also provide greater than 90% power to detect a difference in the change from baseline in HAQ-DI score between treatment groups (two-sided t test on the van der Waerden normal scores, α = 0.05), assuming an improvement from baseline in HAQ-DI of −0.21 for group 1, −0.47 for group 3 and −0.39 for group 4.
Statistical analyses
There were two co-primary endpoints: the proportion of patients achieving an ACR20 response at week 14 and the improvement from baseline in HAQ-DI score at week 24. A two-sided χ2 test was used to analyse the ACR20 data and a two-sided analysis of variance on the van der Waerden normal scores was used to analyse the HAQ-DI data; both were conducted at a significance level of α = 0.05. The co-primary endpoints were analysed sequentially (ACR20 response at week 14 first and HAQ-DI at week 24 second) to maintain an overall type I error rate of 0.05. Additional details regarding statistical testing of the primary endpoint and data handling guidelines are provided in supplemental material 2 (available online only).
RESULTS
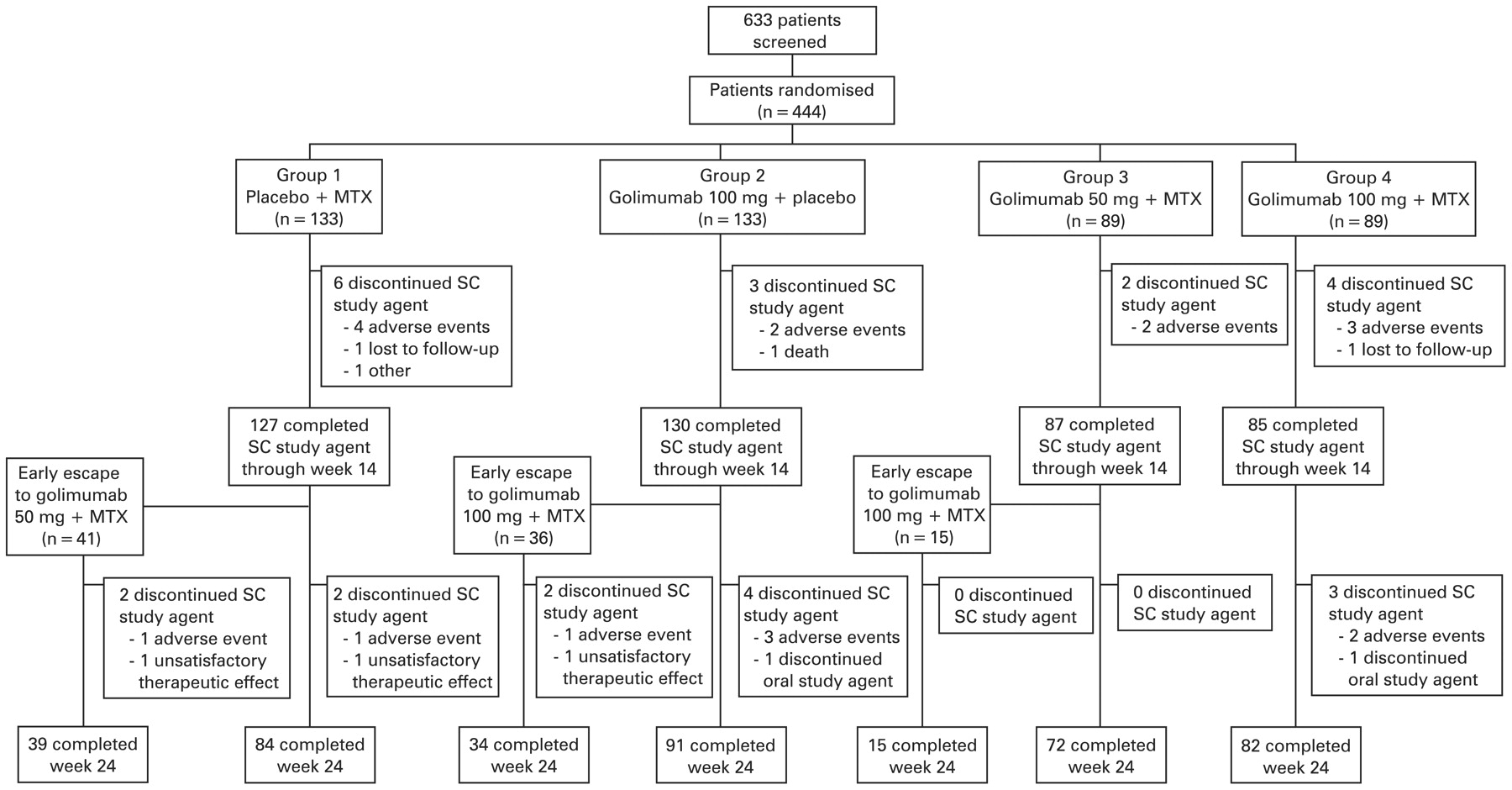
Data were collected between 19 December 2005 and 17 September 2007. A total of 444 patients was randomly assigned to study treatment (fig 1). The treatment groups were generally well balanced with regard to baseline demographic and disease characteristics (table 1).

Disposition of patients during the study. MTX, methotrexate; SC, subcutaneous.
Efficacy
Thirty-three per cent of patients in group 1 achieved an ACR20 response at week 14 compared with 55.6% (p<0.001) in the combined groups 3 and 4, representing 55.1% (p = 0.001) in group 3 and 56.2% (p<0.001) in group 4 (table 2). The proportion of ACR20 responders in group 2 (44.4%) was not statistically significantly different from that of group 1 (33.1%) at the α = 0.05 level (p = 0.059). Four patients (3.0%) in group 1, four patients (3.0%) in group 2, three patients (3.4%) in group 3 and one patient (1.1%) in group 4 were considered not to have achieved the primary endpoint because they met the prespecified treatment failure criteria (see supplemental material 2, available online only).
At week 24, patients in the combined groups 3 and 4 also showed significantly greater improvement in the median HAQ-DI score (−0.44, p<0.001) compared with group 1 (−0.13), with improvements of −0.38 (p = 0.001) in group 3 and −0.50 (p<0.001) in group 4 (table 2). No difference in the improvement in median HAQ-DI score was observed between group 2 and group 1 (−0.13; p = 0.240). Overall, 38.6% of patients in group 1, 45.3% of patients in group 2 (p = 0.276), 68.2% of patients in group 3 (p<0.001) and 72.1% of patients in group 4 (p<0.001) achieved a reduction in HAQ-DI score of 0.25 or greater.
Results of the secondary endpoints supported those of the co-primary endpoints (table 2 and see supplemental table 1, available online only). ACR20 responses were observed in groups 3 and 4 as early as the first assessment at week 4 and generally increased to week 24 (fig 2). The proportions of patients achieving ACR20 responses in groups 1 and 2 peaked at week 14 and subsequently decreased slightly. Significantly greater proportions of patients in the combined groups 3 and 4 also achieved EULAR response, DAS28 remission, and ACR50 and ACR70 responses at weeks 14 and 24 compared with group 1 (table 2).

{kind=link}
{kind=link}
ACR20, ACR50, ACR70 and ACR90 responses to week 24. Patients who entered early escape at week 16 were considered to be non-responders at all subsequent time points. ACR, American College of Rheumatology; MTX, methotrexate.
The co-primary endpoints were evaluated in subgroups of patients divided by geographical region (supplemental table 2, available online only). The proportions of patients with ACR20 responses at week 14 were greater in the combined groups 3 and 4 compared with group 1 in Europe–Australia–New Zealand, North America and Asia but not in Latin America. The placebo response rate in Latin America was greater (68.2%) than that of the other regions (25.4%, 22.6% and 33.3% for Europe–Australia–New Zealand, North America and Asia, respectively). Similar patterns were observed for the improvement from baseline to week 24 in HAQ-DI score. When data from Latin American sites were excluded from the analysis, 26.1% of patients in group 1 had an ACR20 response compared with 41.8% of patients in group 2 (p = 0.014), 55.6% of patients in group 3 (p<0.001) and 54.2% of patients in group 4 (p<0.001).
Overall, 41 patients (31.6%), 36 patients (27.1%) and 15 patients (16.9%) in groups 1, 2 and 3, respectively, met the early escape criteria at week 16 (fig 1). Of these, 17 patients (41.5%), seven patients (19.4%) and three patients (20.0%), respectively, achieved an ACR20 response at week 24. Of the 14 patients (15.7%) in group 4 (100 mg plus methotrexate group) who met the criteria for early escape at week 16 without any dose adjustments, four patients (28.6%) had an ACR20 response at week 24.
Safety
To week 16, before patients had the opportunity for early escape, the proportions of patients with adverse events were 60.9% in group 1, 63.2% in group 2, 68.5% in group 3 and 69.7% in group 4 (table 3). Serious adverse events occurred in 2.3%, 3.8%, 5.6% and 9.0% of patients, respectively.
Safety results to week 24 are presented as the number of events per patient-year to account for patients who entered early escape or discontinued treatment before week 24 (table 4). The numbers of serious adverse events and serious infections per patient-year were greater in group 4 than in groups 1, 2 or 3; however, the 95% CI were wide and overlapping.
One patient died during the study. The patient was assigned to group 2 and received study treatments at baseline and week 4. The patient developed nausea, diarrhoea and dehydration and was hospitalised on study day 56 (week 8), developed an ileus and aspiration pneumonia and subsequently died from sepsis on study day 69 (week 9).
Ninety-two patients (20.7%) were diagnosed at screening with latent tuberculosis but entered the study while receiving treatment for latent tuberculosis (usually isoniazid, table 1). None of these patients, or any other patients, developed active tuberculosis. No patients had opportunistic infections. A total of 12 patients had serious infections to week 24 (table 4), including the previously mentioned patient who died from sepsis.
Four patients had malignancies, including a patient in group 1 with basal cell cancer, a patient in group 2 with squamous cell skin cancer, a patient in group 2 with basal cell cancer and a patient in group 4 with breast cancer.
Injection-site reactions were uncommon (table 3 and table 4), were primarily mild in intensity and consisted most often of injection-site erythema, bruising, or warmth. No injection site reactions were considered to be severe or serious, and no patients discontinued study treatment because of injection-site reactions.
Antinuclear antibodies were observed in similar proportions between the combined groups 3 and 4 (12.2%) and group 1 (14.9%) at week 14, whereas the proportion was greater for patients in group 2 (29.3%) compared with groups 3 or 4 (5.7% and 17.9%, respectively). No patients demonstrated symptoms that would be consistent with lupus-like syndrome.
Overall, five of 236 patients with evaluable samples (2.1%) had antibodies to golimumab at week 24. All of the patients were from group 2. Two of the patients had received golimumab 100 mg plus placebo without early escape and three of the patients had entered early escape to golimumab 100 mg plus methotrexate. Two of the five patients (40%) achieved an ACR20 response at week 24 and one patient (20%) achieved an ACR50 response. None of the patients with antibodies to golimumab had an injection-site reaction.
DISCUSSION
We evaluated the effect of subcutaneous golimumab injections every 4 weeks with or without background methotrexate on the signs and symptoms of RA after 24 weeks in patients with active disease despite treatment with methotrexate. The combination of golimumab and methotrexate was superior to either golimumab or methotrexate alone in improving the signs and symptoms of RA and physical function. This is consistent with the results of previous studies in which anti-TNF agents were evaluated alone or in combination with methotrexate.10 12 No clear difference in the efficacy of the two golimumab dose groups that included concomitant methotrexate was evident. Patients who received golimumab without methotrexate also showed some evidence of benefit (eg, ACR50), but the proportion of patients with an ACR20 response was not statistically significantly greater than that observed for patients who received methotrexate alone.
The baseline characteristics of the study population indicate that patients had moderately to severely active RA. However, patients in this study had shorter disease duration, fewer tender and swollen joints, better physical function and lower CRP levels than those in previous studies of biological agents in patients with active RA despite methotrexate therapy.1 5 10 26 The less restrictive entry criteria in our study (eg, tender and swollen joint counts and CRP) may have allowed for a broader patient population. Recent data suggest that baseline disease activity levels in patients who receive TNFα inhibitors in actual clinical practice have decreased since these drugs first became available.27–29 The lower disease activity in our study cohort may thus be reflective of this overall reduction in the disease activity of patients in actual clinical practice. The shorter disease duration of patients in our study cohort may also reflect an increased tendency to treat patients with anti-TNF agents earlier in their disease course.
Patients who received placebo plus methotrexate (group 1) showed a greater than expected improvement in measures of disease activity relative to previous anti-TNF studies of patients with active RA despite methotrexate treatment.1 8 30 More patients in our study also had exposure to methotrexate for >1 year compared with patients in the previous trials (eg, the ATTRACT study population),1 providing additional evidence that patients are now being treated with anti-TNF therapy earlier in their disease course. Some patients who received methotrexate for shorter durations may not have reached the maximum benefit from methotrexate therapy before entering the study and may have continued to improve during the study, which would have inflated the number of ACR20 responders in group 1. Greater treatment compliance in the context of a clinical trial, in which methotrexate is provided at no cost to patients and doses are monitored and tracked, may also have contributed to this trend. The disproportionately high ACR20 responses (68%) among patients in group 1 from the Latin American countries could also have contributed to this result.
Whereas the response rates with golimumab alone (group 2) seemed to be adequate, clinically relevant and similar to those observed with other anti-TNFα agents alone,30 they were not statistically significantly greater than methotrexate alone in this patient population. The high response rate in group 1 must be considered when interpreting these results. Another contributing factor may have been the unique design of this study that did not include a washout period. A washout period was omitted for clinical and ethical reasons, particularly given the recent advances in treatment strategies of RA and the availability of effective therapies. Patients with active RA would have gone untreated during a washout period, which could have increased their risk of developing irreversible joint damage. All patients in groups 1, 3 and 4 thus continued methotrexate treatment without interruption. For patients in group 2, methotrexate was stopped when golimumab was started and patients received sham methotrexate capsules. Patients in this group whose disease was partly controlled by methotrexate (while still meeting the study entry criteria) would have been likely to experience worsening of disease activity after discontinuing methotrexate. Therefore, this group may have been at a disadvantage compared with the groups who continued methotrexate in terms of showing an improvement in efficacy parameters compared with baseline.
Golimumab was generally well tolerated with no unexpected safety issues, even though many of the study sites were in regions in which tuberculosis is endemic. Approximately 20% of the patients had latent tuberculosis at screening and entered the study receiving isoniazid (or other therapies for latent tuberculosis). None of these patients developed active tuberculosis.
Injection-site reactions were uncommon, occurring in approximately 5% of patients who received golimumab at any time. These reactions were generally mild or moderate and consisted primarily of erythema; none led to discontinuation of the study agent.
Patients in the golimumab groups had greater incidences of infections than those in the methotrexate-alone group. Also, patients in the 100 mg golimumab plus methotrexate group had greater incidences of serious adverse events and serious infections than those in the other groups. One patient who received golimumab 100 mg alone died because of a serious infection. No opportunistic infections or cases of tuberculosis were observed. The observed incidence of antibodies to golimumab was too low to evaluate their effect on clinical efficacy and safety.
In conclusion, the addition of golimumab injections every 4 weeks to methotrexate in patients with active RA significantly reduced the signs and symptoms of RA and improved physical function. The safety profile of golimumab was generally consistent with what is expected for an anti-TNFα agent. However, the study was not powered to assess adverse events; therefore, definitive conclusions about safety cannot be drawn. This study will continue to assess the long-term efficacy and safety of golimumab.
Acknowledgments
The authors would like to thank the patients, investigators and study personnel who made this trial possible. Scott Newcomer, MS of Centocor Ortho Biotech Services LLC, assisted in preparing the manuscript.
REFERENCES
Supplementary materials
Web only appendices 68;6:789-796
Files in this Data Supplement:
Footnotes
Competing interests: Declared. ECK, MCG, LK, STH, PCM, JP, SCB and WP (or their institutions) have received research grants from Centocor and/or Schering-Plough. LK and ECK have received consulting fees from Centocor and/or Schering-Plough. ECH, JZ, SV, ZW and MUR are employees of Centocor Research and Development, Inc and own stock and/or stock options in Johnson&Johnson, Inc. MW is an employee of and owns stock in Schering-Plough Research Institute, Inc.
Funding: This study was funded by Centocor Research and Development, Inc, a wholly owned subsidiary of Johnson&Johnson, Inc and Schering Plough Research Institute, Inc.
The following investigators participated in the GO-FORWARD study: Argentina: Alejandro Alvarellos (Córdoba), Alejandra Magdelena Babinin (Córdoba), Juan Carlos Barreira (Cuidad Autonoma de Buenos Aires), Alberto Berman (Tucman), Maria Alicia Lázaro (Buenos Aires), Ingrid Strusberg (Córdoba), Guillermo Alberto Tate (Buenos Aires), Osvaldo Hübscher (Cuidad Autonoma de Buenos Aires), Bernardo Antonio Pons Estel (Santa Fé); Australia: Stephen Hall (Malvern, VIC), Geoffrey Owen Littlejohn (Clayton, VIC), Peter Thomas Nash (Maroochydore and Cotton Tree, QLD); Canada: Christopher Atkins (Victoria, BC), André Beaulieu (Ste-Foy, QC), Alfred Cividino (Hamilton, ON), Edward Keystone (Toronto, ON), Majed Khraishi (St John’s, NL), Robert McKendry (Ottawa, ON); Chile: Pedro Claudio Miranda Cabezas (Santiago), Ana Flores Torterolo (Rancagua), Irmgadt Annelise Goecke (Santiago), Maria Loreto Massardo Vega (Santiago), Juan Pablo Riedemann Gonzalez (Temuco); Germany: Holm Häntzschel (Leipzig), Rolf W Hauer (Berlin), Gunther Neeck (Rostock), Andrea Rubbert (Köln), Jürgen Wollenhaupt (Hamburg), Reinhold Ernst Schmidt and Henning Zeidler (Hanover); Hungary: László Hodinka (Budapest); Mexico: Jorge Antonio Esquivel Valerio (Monterrey, NL); New Zealand: Daniel Wai Tho Ching (Timaru), Nicola Dalbeth (Auckland), Peter Brian Barrie Jones (Rotorua); Poland: Jan Brzezicki (Elblag), Marek Stefan Brzosko (Szczecin), Zbigniew Adam Mencel (Kalisz), Anna Fillipowicz-Sosnowska (Warszawa), Hanna Chwalińska-Sadowska (Warszawa), Jacek Andrzej Pazdur (Warszawa); South Korea: Sang Cheol Bae (Seoul), Hyun Ah Kim (Kyunggi-do), Sung II Kim (Seo-Gu), Choong Ki Lee (Nam-gu Daegu), Soo Kon Lee (Seoul), Yeong Wook Song (Seoul); Republic of China: Tien-Tsai Cheng (Taiwan); USA: Antony Hou and Eugene P Boling (Upland, CA), Richard D Brasington, Jr (St Louis, MO), Melvin A Churchill, Jr (Lincoln, NE), Gino DiVittorio (Mobile, AL), John E Ervin (Kansas City, MO), Mark Genovese (Palo Alto, CA), Mitchell Lowenstein (Palm Harbor, FL), Michael Miniter (Moline, IL), Frederick T Murphy (North Duncansville, PA), William Palmer (Omaha, NE), Irene Tong (Pasadena, CA), Yong Tsai (Ormond Beach, FL), Christopher Wise (Richmond, VA).
▸ Additional supplemental material is published online only at http://ard.bmj.com/content/vol68/issue6
Ethics approval: The protocol was reviewed and approved by each site’s institutional review board or ethics committee.
Patient consent: Obtained.
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.