Article Text

Abstract
Background: Mesenchymal stem cells (MSCs) have a potential immunomodulatory role in autoimmune disease; however, the qualitative properties and haematopoietic support capacity of MSCs derived from patients with autoimmune disease is unclear.
Objectives: To further characterise phenotypically and functionally bone marrow (BM)-derived MSCs from patients with systemic sclerosis (SSc).
Methods: Key parameters of BM-derived MSC function and phenotype were assessed in 12 patients with SSc and compared with 13 healthy normal controls. The parameters included the ability to: form colony-forming unit fibroblasts (CFU-F), differentiate along the adipogenic and osteogenic lineages, express cell surface antigens defining the MSCs population, support normal haematopoiesis and suppress in vitro lymphocyte proliferation induced by either anti-CD3∊ plus anti-CD28 monoclonal antibodies or the mixed lymphocyte reaction.
Results: SSc MSCs were shown to have a similar characteristic phenotype, capacities to form CFU-F and to differentiate along adipogenic and osteogenic lineages as those of healthy donor MSCs. The ability of SSc MSCs to support long-term haematopoiesis was also identical to that of controls. Both healthy donor and SSc BM MSCs reduced the proliferation of autologous and allogeneic peripheral blood mononuclear cells in a cell number dependent fashion.
Conclusions: These results show that BM-derived MSCs from patients with SSc under the described culture conditions exhibit the same phenotypic, proliferative, differentiation potential and immunosuppressive properties as their healthy counterparts and could therefore be considered in an autologous setting. Further studies are needed to ensure the quality and safety of large-scale expansion of patient MSCs prior to their potential use in clinical trials.
Statistics from Altmetric.com
The bone marrow (BM) microenvironment contains a complex network of non-haematopoietic cells, mainly of mesenchymal origin, which provide support for growth, proliferation and differentiation of the haematopoietic stem cells and their progeny. Within these cells, a population of multipotential progenitors, referred to as mesenchymal stem cells (MSCs) or, more correctly multipotent mesenchymal stromal cells1 has been identified. MSCs may also be isolated from adipose tissue, fetal liver, cord blood or the synovial membrane.2 MSCs are capable of differentiating along bone, cartilage, muscle and fat lineages.3 These cells are identified in vitro by their ability to form colony-forming unit fibroblasts (CFU-F) and, phenotypically, by their expression of surface markers, such as CD105, CD73, CD90, integrins and adhesion molecules in addition to the absence of CD14, CD34 and CD45 haematopoietic-related antigen.4 5 MSCs have also been shown to exhibit immune modulatory functions. In vitro, human MSCs inhibit or suppress several T cell functions.6–10 MSCs induce T cell division arrest in mixed lymphocyte reactions (MLR); this effect is irreversible upon removal of the MSCs in murine models. In contrast to the strong inhibitory effects of MSCs on T cell proliferation, there are only relatively minor and reversible effects on the T cell effector function as measured by interferon (IFN) γ production, and no effects on T cell activation, as assessed by CD25 and CD69 surface expression. The T cell inhibition does not appear to be antigen specific, it operates across HLA barriers and targets primary and secondary responses.
MSCs can also inhibit B cell proliferation and differentiation,11 dendritic cell maturation and proliferation,12–14 and suppress natural killer cell proliferation and cytokine production, such as interleukins 2 and 15.15 16 The immunomodulatory mechanisms are multiple, involving both cell contact and soluble factors (eg, indoleamine 2.3 dioxygenase, transforming growth factor β, prostaglandin E2), and were recently reviewed.17
In vivo, MSCs are proposed to give rise to the majority of marrow stromal cell lineages and to participate in regulating the balance between haematopoietic stem cell self-renewal and differentiation.18 Based on these important properties, several recent clinical trials have been designed to use MSCs either to enhance donor haematopoietic stem cell engraftment19 or to exploit their antiproliferative and anti-inflammatory properties to treat or prevent acute graft versus host disease (aGvHD) in allogeneic haematopoietic stem cell transplant recipients.20 21 Recently, the potential putative and/or therapeutic potential of MSCs are being studied in autoimmune disease (AD). Current AD experimental models are inconsistent. Syngeneic MSC injection in mice with experimental autoimmune encephalomyelitis successfully prevented or ameliorated the disease progression.10 22 However, in one study in a mouse model of collagen-induced arthritis, MSCs exacerbated the disease23 while they ameliorated the disease in another study,24 thus demonstrating the heterogeneity of AD diseases.
In people with aGvHD who were heavily immunosuppressed, only allogeneic MSCs have been employed, and an apparent immune privilege has been demonstrated. However, in non-immunosuppressed mice, allogeneic MSCs were rejected.25 A better characterisation of human AD MSCs is therefore a critical prerequisite for their therapeutic use in an autologous setting. However, some data suggest that BM MSCs derived from patients with AD (rheumatoid arthritis, systemic lupus erythematosus and systemic sclerosis (SSc)) are defective in their differentiation and haematopoiesis stromal support functions.26–28
In this study, we investigated in vitro some key functions of BM MSCs derived from 12 patients with systemic sclerosis, some of whom having undergone autologous haematopoietic stem cell transplantation following intensive immunosuppression. The engraftment experiences of these patients act as an in vivo surrogate marker of stromal function.
MATERIALS AND METHODS
Patients
Bone marrow samples were obtained from 12 patients with SSc, of which nine had early rapidly progressive diffuse cutaneous SSc (dcSSc) and three the limited cutaneous (lcSSc) subsets.29 BM aspirate was obtained either via a sternum aspirate (eight patients, Saint-Louis Hospital) or via the iliac crest (four patients, University Hospital Basel). Two patients had undergone haematopoietic stem cell transplantation for the treatment of rapidly progressive dcSSC. Patients’ disease and clinical characteristics are shown in table 1. As controls, BM samples of healthy donors were obtained either from nine filters used during BM processing for allogeneic transplantation or from four healthy donors undergoing spinal surgery. Sternum-derived aspirates yielded smaller volumes and a reduced number of expanded BM MSC, which limited the number of tests possible to perform on single specimens. Approval from local ethics committee and signed healthy donor/patient consent were obtained.
Cell culture
BM mononuclear cells from patients and healthy donors were isolated by Ficoll (Invitrogen, Cergy-Pontoise, France or Ficoll-Hystopaque, density 1.077 g/l, Sigma-Aldrich Inc., St Louis, MO, USA) gradient and cultured at an initial density of 5×104 cells/cm2 in minimum essential medium-α (Invitrogen), supplemented with 10% defined fetal calf serum (HyClone, Logan, UT, USA), l-glutamine (2 mM; Invitrogen), antibiotic/antimycotic (Invitrogen) and basic fibroblast growth factor (bFGF or FGF-2) (1 ng/ml; R&D Systems, Lille, France). Some BM MSCs from patients with SSc and healthy controls were alternatively expanded in α-modified MEM medium supplemented with 10% fetal bovine serum (both from Gibco-BRL, Carlsbad, CA, USA) as already described30 with the addition of bFGF. After 24–48 h, non-adherent cells were removed and medium was changed. Cultures were fed every 2 or 3 days until confluence. Adherent cells were then trypsinised, harvested and subcultured by seeding 1×103 cells per cm2. Most assays were performed on cells within the third expansion passage, mostly at the first and second. Mycoplasma contamination was tested by using Venor GeM Mycoplasma Detection Kit (Minerva Biolabs, Berlin, Germany) in selected cultures (see Results) and were negative.
Characterisation of mesenchymal stem cells
The median number of CFU-F per 106 nucleated cells was calculated by plating Ficoll gradient purified mononucleated cells from healthy controls (n = 4) and patients with SSc (n = 4) at a density of 4500 cells/cm2. Colonies were counted 2 weeks later.
For surface phenotype analysis, MSCs were stained with a core set of antibodies directed to CD34, CD45, CD73, CD90, CD13, Cd49a CD44, CD105, HLA-AB and HLA-DR or matched isotype control (all purchased from Becton Dickinson, Le Pont de Claix, France). Immunofluorescence analysis was performed by use of a five-parameter flow cytometer (FACScalibur, Becton Dickinson, San Jose, CA, USA).
Osteogenic and adipogenic in vitro differentiation of MSCs cells was performed according to published protocols30 31 on cells at their first or second passage.
In vitro peripheral blood mononuclear cell proliferation assay with anti-CD3∊ monoclonal antibodies
The proliferation of peripheral blood mononuclear cells (PBMCs) from healthy donors and patients with AD in the presence of BM MSCs was performed as previously described.30 Irradiated (30 Gy) BM MSCs cells were seeded at dilutions (each in triplicate) of: 0.5, 2, 10 or 50×103 cells per well and allowed to attach for at least 1 h at 37°C before adding PBMCs.
PBMCs (105 per well) in RPMI 1640 medium, supplemented with 5% pooled human serum, were added with or without anti-CD3∊ monoclonal antibody (0.5 μg/ml, a gift from Antonio Lanzavecchia, Bellinzona, Switzerland) and anti-CD28 monoclonal antibody (1 μg/ml, Becton Dickinson, USA) The plates were incubated at 37°C for 48 h, then pulsed for 18 h with 1 μCi/well 3H-thymidine (Amersham, Les Ulis, France), harvested and the 3H cpm counted.
Mixed lymphocyte reactions
PBMCs from a normal donor were mixed with patients that had been irradiated (25 Gy) or control MSCs and PBMCs from another HLA-DRB mismatched healthy individual, and resuspended in RPMI-1640 medium supplemented with 10% defined fetal calf serum (Hyclone), Hepes (25 mM), l-glutamine (2 mM), penicillin, streptomycin (Gibco-BRL). PBMCs were seeded in triplicate at a concentration of 1×105 cells/50 μl/well. RPMI (50 μl) or MSCs at various concentrations (from 0.1 to 200×103 MSCs per well) were added in the wells at the beginning of the experiment. MSCs were allogeneic to responder and stimulatory PBMCs. After 5 days of incubation, 1 μCi of 3H-thymidine (Amersham) was added overnight and thymidine incorporation was measured using a β-scintillation counter (Beckman, Gagny, France) and expressed as cpm.
Mesenchymal stem cell-mediated support of haematopoiesis
MSCs were irradiated (25 Gy) and subcultured at a concentration of 3×104 MSCs per well as previously described.31 The plates were recharged with 100, 200 or 500 CD34+ cells/well and incubated for 5 weeks with weekly culture refeeding. Fifteen replicate wells per dilution were established. Weekly, the entire content of three wells of each dilution was harvested: non-adherent and adherent cells were plated into 24-well with 0.5 ml/well of complete methylcellulose medium (Stem alpha, Saint Clement les Places, France) and maintained at 37°C in 5% CO2. Colony forming cell (CFC) quantification in each well was evaluated at day 14.
Statistical analysis
The Student t-test for paired data was used to test the probability of significant differences between samples. A value of p<0.05 was used to define statistical significance.
RESULTS
Characterisation of bone marrow mesenchymal stem cells
BM MSCs from patients with SSc (n = 12) and healthy donors (n = 13) showed no significant differences in morphology or in proliferation rate in the culture conditions used containing, in addition to fetal bovine serum, also 1 ng/ml of bFGF (fig 1A).

This BM MSC expansion regimen has been chosen in French trials to treat acute GvHD (Sensebe L, personal communication). The use of bFGF has been shown to improve cell proliferation and not to alter their basic in vitro functions.32 33 The effect of bFGF, though at 1 ng/ml concentration, significantly improves proliferation and morphology of all cultured MSCs, healthy or SSc.
Colony-forming unit fibroblast formation
CFU-F formation was tested on four SSc and four healthy donor BM MSCs. The median CFU-F number among SSc and healthy donors MSC grown in the presence of bFGF, was 91.5 (range 6–124) and 76 (22–157) respectively. When the same SSc and healthy BM MSC were grown in the absence of bFGF, 10–20% more CFU-Fs but with a smaller size and lower staining intensity were obtained as reported already in the literature.32 (Bocelli Tyndall, unpublished data).
Immunophenotype
Phenotypically, SSc MSCs highly expressed CD73, CD90, CD13, CD105, HLA-ABC, integrin (CD49e) and adhesion molecules (CD44) and were mostly negative for HLA-DR expression (table 2). The absence of contaminating haematopoietic stem cells was confirmed by the negative expression of CD34 and CD45 antigens in all studied cases and in five cases also for CD14 antigen. No phenotypic differences were observed between BM MSCs from patients with SSc and controls. It must be, however, pointed out that HLA-DR was significantly expressed in two controls and two SSc BM MSCs. One SSc sample showed expression of HLA-DR on 79% of the total cells and the other 23.4%. The same cells, however, had insignificant (2%) expression of HLA-DR when grown without bFGF (Bocelli Tyndall, unpublished data). Similarly, two controls were HLA-DR positive (76% and 10% of the cells) when grown with bFGF. The expression of HLA-DR was not due to mycoplasma contamination. It has been reported also by others that BM MSCs grown in the presence of 1 ng/ml bFGF can express HLA-DR.34
In vitro differentiation of mesenchymal stem cells from patients with systemic sclerosis
To assess the adipogenic and osteogenic potential of BM-derived SSc MSCs, cells were expanded and cultured in standard induction medium. All 12 SSc and 10 healthy BM MSCs have been tested and all differentiated toward the osteogenic and adipogenic lineages. Morphological changes and formation of lipid-rich vacuoles within the cells were observed in both patient and control MSCs (fig 1B). Lipid droplets were noticeable after about 2 weeks of induction, the amount and volume of droplets increasing thereafter in a time-dependent manner. SSc MSCs were also shown, like control MSCs, to differentiate into osteocytes as demonstrated by an upregulation of alkaline phosphatase activity (fig 1B). These findings suggest that adipogenesis and osteogenesis potential of MSCs from patients with SSc are qualitatively conserved. However, within each group of SSc and healthy BM MSCs variations of differentiation efficiencies in each lineage occurred, as measured by the qualitative colorimetric assessments, without any obvious specific pattern.
Effect of mesenchymal stem cells from patients with systemic sclerosis on peripheral blood mononuclear cell proliferation
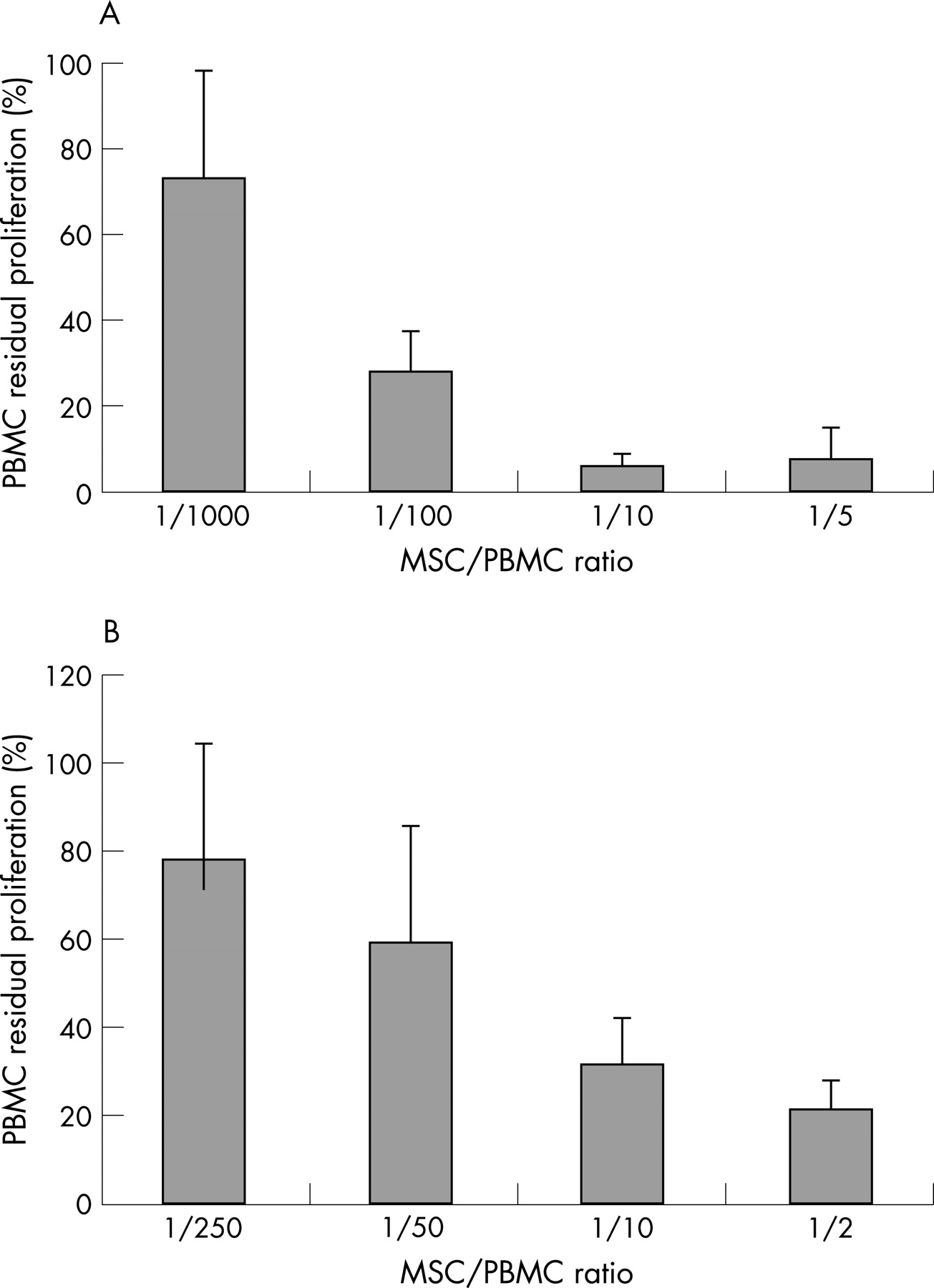
The effect of patient and control MSCs on PBMC proliferation was evaluated in MLR assays or on anti-CD3∊-stimulated PBMCs. In MLR, MSCs and allogeneic PBMC were mixed at four different ratios (1:1000, 1:100, 1:10, 1:5). MLRs were performed with MSCs from three patients with SSc and five controls. SSc MSCs inhibited PBMC proliferation in a cell-dependent manner with less than 10% residual proliferation for ratio 1:5 (fig 2A). The antiproliferative effect of SSc MSCs was still effective at lower concentration as the measured PBMC residual proliferation was as low as 27% at ratio 1:100. No significant differences in MSC-mediated inhibition of proliferation were observed between patients with SSc and controls.

The effect of MSCs on PBMC stimulated to proliferate with anti-CD3∊ (and anti-CD28) as described in Methods, was assessed on four SSc and two control BM MSCs (different from those assessed above with MLR). MSC to PBMC ratios were 1:250, 1:50, 1:10 and 1:2. The PBMCs were either autologous or allogeneic to the SSc MSCs.
The results are summarised in fig 2B. SSc MSCs reduced the proliferation of both allogeneic and autologous PBMC up to almost 90% at the highest MSC to PBMC ratio (1:2) and at least to 50% at the 1:10 ratio, without any significant difference among themselves or the controls MSCs. For this reason all results are presented together. These results are similar to those already published on other control and AD BM MSC, which had been grown without the addition of bFGF30 and are the same as those obtained with the SSc MSC in this study grown without bFGF (Bocelli-Tyndall, data not shown). In MLR assays, the reduction of the PBMC proliferation by the MSCs seems even more pronounced than with anti-CD3∊ stimulation, but these differences are more likely to reflect different experimental conditions and assays.
Long-term haematopoiesis support
The capacity of MSCs to support normal long-term haematopoiesis was assessed for nine patients with SSc and nine controls. Irradiated MSC layers were recharged with three different concentrations of CD34+ cell isolated from healthy donors (100, 200 or 500 CD34+ cells). The experiments were performed at the same time and with the same CD34+ cells for both patients with SSc and control MSCs. As shown in fig 3, the number of CFCs obtained weekly over a period of 5 weeks did not differ significantly between patients and control subjects, whatever the CD34+ cells concentration tested. In addition, a dose-dependent relationship between the number of inoculated CD34+ cells and the number of CFCs at the end of the culture was observed in both groups. These results strongly suggest that SSc MSCs efficiently support the growth of normal haematopoietic stem cells.

{kind=link}
{kind=link}
{kind=link}
DISCUSSION
This study describes some phenotypic and functional features of in vitro expanded BM MSCs from 12 patients with SSc. Although for technical reasons not all samples could be tested in every assay, the consistency of the data allows some conclusions. The cells present the expected immunophenotype, including the already described34 occasional expression in both SSc and control cells of the HLA-DR antigens in the culture conditions used (containing bFGF).
The SSc BM MSCs have CFU-F ability with a phenotype and frequency similar to that of MSCs derived from healthy donors. They differentiate into adipocytic and osteogenic cells with variabilities, at least on the basis of the colorimetric assays used, similar to those observed within the control healthy BM MSCs. This is different from other reports in which complete failure to differentiate into osteogenic and adipogenic lineages were described.28 However, in this study the expanded MSC cells were derived from a low affinity nerve growth factor binding cell population, which supposedly represent a more stem cell enriched population. The BM MSCs used in our study include several heterogeneous populations likely to be already at different degrees of differentiation.
We show for the first time that, in all cases tested and independently of the disease status, SSc MSCs support in vitro long-term haematopoiesis from added allogeneic healthy CD34+ cells as efficiently as control MSCs, and are equally effective as control healthy donor MSCs in inhibiting in vitro allogeneic and autologous PBMC proliferation.
This is important regarding the potential therapeutic application where an infusion of expanded autologous MSCs may be safer and ethically more acceptable. In establishing the functions of the BM MSCs in AD, a correlation of the in vitro results with the in vivo experience should be included when possible. To date, few preliminary data on BM haematopoietic stem and stromal cells have been reported. In patients with systemic lupus erythematosus, a decreased number of haematopoietic stem cells and a diminished efficiency of MSCs to support haematopoiesis have been reported.26 Similarly, in patients with rheumatoid arthritis, a defect in the MSC-mediated support of haematopoiesis was attributed to an increased production of tumour necrosis factor α.27 The same group has shown that stromal cells from patients with multiple sclerosis display instead normal in vitro haematopoiesis supporting capacity.35 These in vitro data question the feasibility and suitability of such patients with AD to undergo autologous stem cell (CD34+) transplantation. In this procedure the reinfusion of autologous CD34+ cells, mostly derived from the peripheral blood, into the immunoablated SSc host leads to new haematopoiesis and immune reconstitution; this requires correct functioning of both the repopulating CD34+ cells and of the BM stromal cells, from which the MSCs are derived. However, since then many hundreds of patients have received an autologous haematopoietic stem cell transplantation as treatment of an AD, with no suggestion of failed or significantly delayed haematopoietic engraftment. Currently, the published literature on patients with systemic lupus erythematosus, rheumatoid arthritis and multiple sclerosis transplanted with autologous CD34+ stem cells, shows that both neutrophil and platelet engraftment were within the expected normal range in all patients with systemic lupus erythematosus,36 37 rheumatoid arthritis38 and multiple sclerosis.39 Concerning SSc, our results show that the haematopoiesis supporting capacity of MSCs in patients with SSc did not differ from that of normal controls. The CFC numbers were similar in SSc and healthy long-term BM stromal cultures supplemented with healthy CD34+ cells. These data are also consistent with the results obtained in vivo in patients with SSc who have received autologous CD34+ transplantation (as did some of the patients in this study cohort) and did not experience any graft failure or delayed haematopoietic recovery.40–42 These in vivo results are indicative of correct functioning of both stromal (MSC) and haematopoietic CD34+ cells in patients with SSc.
The BM MSC cells in SSc have an antiproliferative effect on allogeneic and autologous proliferating PBMC as healthy controls, extending the results already published30 thus supporting potential autologous clinical application. This “normal” in vitro antiproliferative potential of the BM MSCs in SSc theoretically allows for their use in autologous reinfusion as immunosuppressants as previously discussed.
However, a problem so far not addressed is that of the in vitro expansion of the BM MSCs in SSc to a sufficiently high number within limited passages. The use of bFGF at a low concentration (1 ng/ml), would allow such expansion and seems to prevent, even at the low concentration used, some premature senescent morphology and reduced proliferation rate that we have experienced in culturing both healthy and SSc BM MSCs in media supplemented only with fetal bovine serum as growth adjuvant (Bocelli-Tyndall, unpublished data). However, as shown also by our data, cells from some patients grown with bFGF could be induced to express HLA-DR antigens for reasons that are unclear. However, the MSCs with high HLA-DR expression do not induce any antigenic response in vitro in allogeneic PBMC as shown by others33 34 and the problem could perhaps be negligible if the cells were used in an autologous setting.
Therefore if autologous BM MSCs are contemplated as therapeutic agents in severe therapy resistant SSc or other indications, our data suggest that they are equally effective as normal healthy cells in both haematopoietic support and antiproliferative action. In addition the use of low-dose bFGF prevented premature senescence, improved expansion dynamics and the expression of class II surface antigens was not associated with antigenicity.
Acknowledgments
We would like to thank Dr R Gosert, Institute for Medical Microbiology Transplantation Virology, University of Basel, for performing the mycoplasma detection assays and Marie Noëlle Lacassagne and Brigitte Ternaux, Cell Therapy Unit, Saint Louis Hospital, for their technical assistance.
REFERENCES
Footnotes
Competing interests: None.