Article Text

Abstract
Background: Vascular damage is a key pathological process in systemic sclerosis (SSc) and accounts for significant disease-related morbidity. To determine the clinical burden of severe digital vasculopathy (SDV), we have reviewed hospital-based treatment for this important complication of SSc in a large single centre cohort.
Methods: Cases were identified from a cohort of 1168 patients with a diagnosis of SSc who were reviewed during an 18-month period. Patients with recorded episodes of SDV-related complications (digital ulceration, critical digital ischaemia or digital gangrene), requiring surgical amputation, digital sympathectomy or admissions for intravenous prostacyclin or calcitonin gene related peptide (CGRP) and/or intravenous antibiotic treatment were identified.
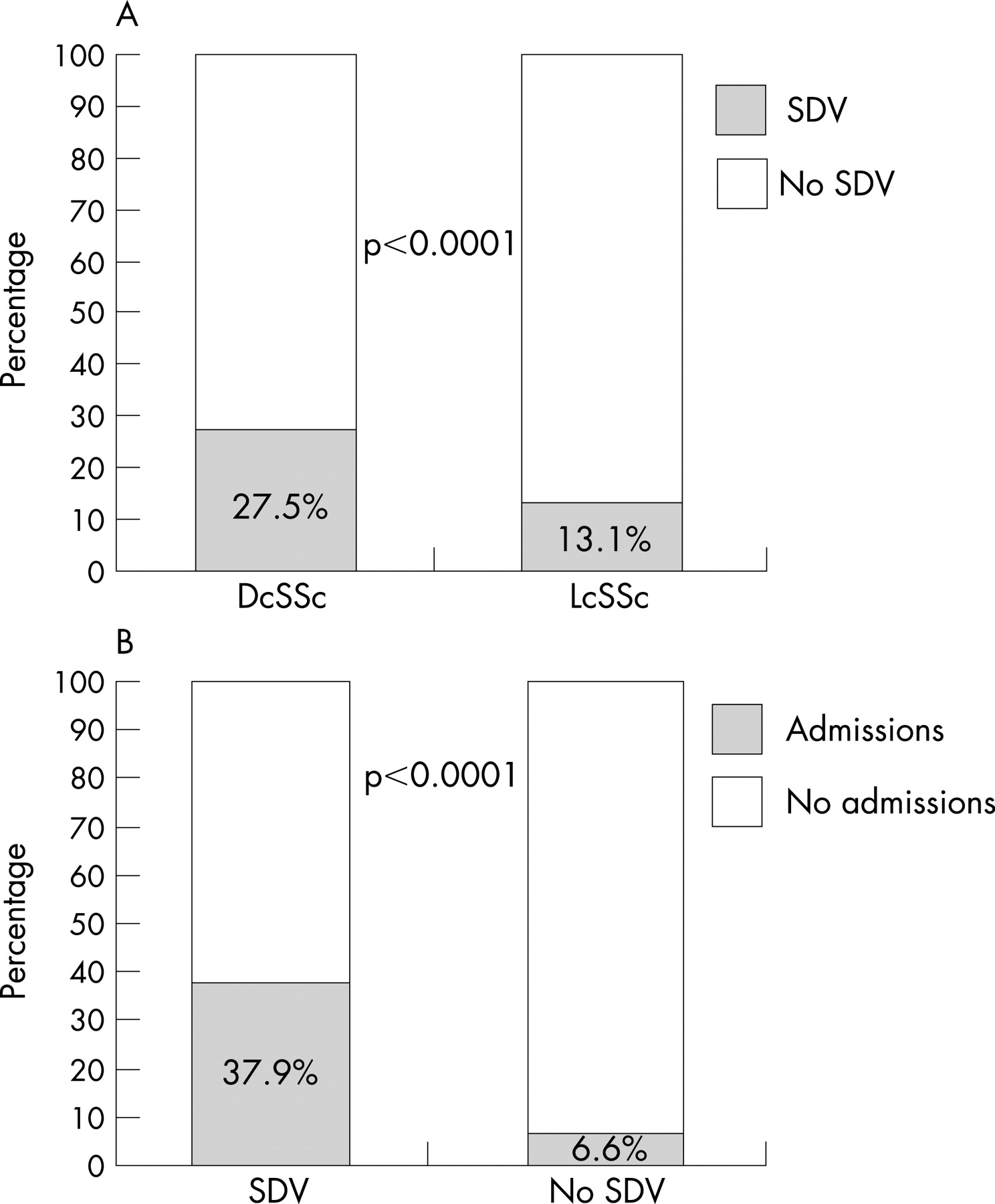
Results: From this large SSc cohort, 17.4% had SDV-related complications. Contrary to expectation, their frequency was significantly higher among the patients with the diffuse cutaneous subset of SSc (27.5%) compared with 13% among the patients with limited cutaneous SSc (p<0.0001). 16.6% had at least one recorded episode of digital ulcers, and 12% required at least one hospitalisation during the 18 months for treatment with intravenous prostacyclin/CGRP. Overall, there were 242 admissions with a mean duration of 6 days.
Conclusions: Digital vasculopathy is a serious complication of SSc contributing significant morbidity and often requiring hospital-based management.
Statistics from Altmetric.com
Systemic sclerosis (SSc) is driven by three interdependent pathophysiological processes: immunological activation, tissue fibrosis and vasculopathy. The most frequent external manifestation of vasculopathy in scleroderma is digital ulceration (DU). Secondary infection, particularly in immunosuppressed patients, may complicate ulceration and necessitate hospital-based treatment.
Ferri et al found the overall frequency of DU among 1012 Italian patients to be 48% and 54% at the beginning and end of follow-up, respectively.1 More recently, the UK Raynaud’s and Scleroderma Association published the results of a self-reported scleroderma questionnaire demonstrating that 46% of the respondents had suffered from DU at some point in their disease history.2
Although many authors have published data on the frequency of DU in patients with SSc over the course of their disease, there is little published information about incidence and prevalence of SDV-related complications and the extent of medical intervention those have required. To determine the clinical burden of severe digital vasculopathy (SDV), we have reviewed the incidence of SDV-related complications over an 18-month period in a large single centre cohort of SSc cases.
METHODS
Cases of SSc affected by SDV-related complications were ascertained, and demographic and clinical characteristics were recorded. SDV was defined as Raynaud’s phenomenon complicated by DU, critical digital ischaemia or gangrene, or requiring digital sympathectomy. DU was defined as an area of loss of surface epithelialisation affecting the digital pulp or bony prominences, not including fissures or areas of calcium extrusions. Critical digital ischaemia was defined as a sustained reduction in digital perfusion with impaired tissue viability, while digital gangrene was considered when the above changes had caused significant tissue loss. In addition, all patients with SSc who were hospitalised in order to receive intravenous prostacyclin/calcitonin gene related peptide (CGRP) for treatment or prevention of SDV were identified.
RESULTS
During an 18-month period, 1168 patients with confirmed diagnosis of SSc were reviewed. Of these, 29.5% (n = 345) had diffuse (dcSSc), and 70.5% (n = 823) had limited cutaneous SSc (lcSSc). Among these, we identified 203 patients (17.4% of the SSc cohort) with SDV. Table 1 summarises the demographic and clinical characteristics of the SDV cohort.
The median (SD) duration of their disease calculated at the middle of the 18-month period was 104 (102) months (range from 7 to 500 months). Of the 203 SDV patients, 53.2% (n = 108) had lcSSc. Considering the structure of the cohort from which cases were recruited, the frequency of SDV among the patients with dcSSc was significantly higher than that among the lcSSc patients (27.5% and 13.1%, respectively, p<0.0001) (fig 1A). Also, 4.4% (n = 9) of the SDV patients had a coexisting macrovascular disease (atherosclerosis in 8 cases and Buerger’s disease in 1 case).

One-third (n = 65) of the SDV cohort was on active immunosuppressive treatment. Three-quarters of the patients with SDV were receiving at least one vasodilator (47.3%, 1 vasodilator; 24.1%, 2 vasodilators; and 3.4%, 3 vasodilators). These included calcium channel blockers, angiotensin converting enzyme inhibitors, angiotensin II receptor blockers, selective serotonin reuptake inhibitors and glyceryl trinitrate. In addition, 18.7% (n = 38) of the patients were on antiplatelet agents (25 on aspirin and 14 on clopidogrel).
Figure 2 describes the frequency of the different SDV-related complications in our cohort. We found 194 patients (16.6% of the SSc cohort) with at least one recorded episode of DU. The average number of DU episodes over the 18-month period in these patients was 1.6 (1), while the average number of ulcers they had was 2.6 (2) (figs 2B, C). Of the 194 patients, 184 had finger ulcers, and 18 had toe ulcers (15.8% and 1.5% of the SSc cohort, respectively). 19 patients (1.6%) developed critical digital ischaemia, and 16 (1.4%) developed digital gangrene. In 11 (0.9%) patients, this resulted in auto- or required surgical amputation, and 5 (0.4%) of the patients had a history of amputation prior to the 18-month follow-up period. 13 patients (1.1%) were referred for digital sympathectomy.

{kind=link}
{kind=link}
Of the 1168 patients, 12.1% (n = 141) were admitted to hospital at least once to receive intravenous prostacyclin or CGRP during the 18-month window. Of these, 77 had at least one recorded episode of SDV-related complication, and the remaining 64 were receiving the treatment as a preventive measure. In the whole SSc cohort, the frequency of admissions was significantly higher among the SDV patients (37.9%, n = 77), compared with patients with no recorded SDV episodes (6.6%, n = 64) (p<0.0001) (fig 1B). Sixty-three patients (5.4% of the SSc cohort) required multiple admissions. There were a total of 242 admissions with a mean duration of 6 days, and each patient was hospitalised on average twice, which accounted for a total of 1346 bed-days (9.5 days in hospital per patient over the 18 months on average). In addition, 18 patients (1.5% of the SSc cohort) required parenteral and 56 (4.8%) oral antibiotic treatment for infected digits, and 20 (1.7%) needed opioid analgesics for SDV-related pain.
DISCUSSION
Our findings confirm the substantial clinical burden of digital vascular disease in SSc. We reviewed frequency of SDV-related complications in a retrospective cohort of 1168 SSc patients and analysed patterns of hospital admissions and treatment. We found that 17.4% of our patients had at least one episode of SDV-related complication, and 12.1% of the cohort required hospital admissions for treatment with intravenous prostacyclin/CGRP as a therapeutic or preventive measure against SDV-related complications during a time window of 18 months.
Previous studies have reported DU in more than 50% of the SSc patients. Robust data are available from several recently reported prospective clinical trials. Among the participants in the RAPIDS-1 study, which explored the prevention of development of new digital ulcers in SSc patients treated with bosentan, 58% of the actively treated patients and 61% of the patients on placebo developed new DUs during the 16 weeks of the study.3 The patients receiving bosentan developed on average 1.4 new ulcers per patient compared with 2.7 in the placebo group. In another large study comparing frequency of digital ulcers among 213 patients randomised to quinapril or placebo and followed up for 2–3 years, 40% developed DUs during the course of the study.4 In our cohort, less than 17% of the patients developed DUs over the 18-month period with an average number of DUs per patient being 2.6. The lower frequency of DU observed in our study may be explained by the relatively short time period of evaluation. Furthermore, the gap between reviews varied widely between patients with those with otherwise stable disease often being seen once every 15–18 months. It is likely that episodes of DU that are not present at clinic attendance were under-reported. Additionally, uncomplicated digital ulcers which do not require any specific treatment are perhaps also less likely to be recorded by the clinicians during routine evaluation causing potential bias towards more severely affected cases.
It is generally believed that vascular manifestations are more prominent in patients with lcSSc.5 Interestingly, in our cohort with SDV patients, the two subsets were almost equally represented (47% diffuse and 53% limited). Considering the distribution of the two major subsets of SSc in our cohort, we were surprised to find out that the frequency of SDV among the dcSSc patients is significantly higher than that among the lcSSc patients (27.5% and 13%, respectively), which has been observed previously by other authors.6 One potential explanation for this rather surprising finding includes bias towards dcSSc because these cases are seen more frequently, so ascertainment of ulcers may be more complete over the 18-month study window. Future prospective studies are needed to address this point.
Our findings provide some information about the clinical and laboratory features of SSc cases that are most strongly associated with complications of digital vasculopathy. The most frequently seen autoantibodies in our SDV cohort were the antitopoisomerase antibody followed by the anticentromere antibody. This supports the concept that more severe complicated digital vasculopathy is associated with scleroderma-specific autoantibodies.7 8 The disease duration among our patients varied between 7 months and more than 40 years, and there was no significant correlation between disease duration and SDV.
A major limitation of our study is the retrospective design which inevitably led to some missing data and probably to an underestimation of the frequencies of SDV-related complications. In addition, some patients are under shared care, which led to an underestimation of the frequency of hospitalisations and hospital-based and ambulatory treatment. However, unlike other authors, we looked at a fixed short time period, which allowed us to calculate the incidence of SDV-related complications and analyse the clinical burden of this serious SSc manifestation in terms of hospital admissions and treatment requirements.
In conclusion, we have demonstrated that SDV is a serious complication of SSc contributing to a significant morbidity and often requiring inpatient hospital management.
Acknowledgments
This study has been funded by an unrestricted research grant from Actelion pharmaceuticals. In addition, all research projects in our department benefit from support from Arthritis Research Campaign (ICAC grant reference 17692) and Raynaud’s and Scleroderma Association.
Footnotes
-
Competing interests: None declared.